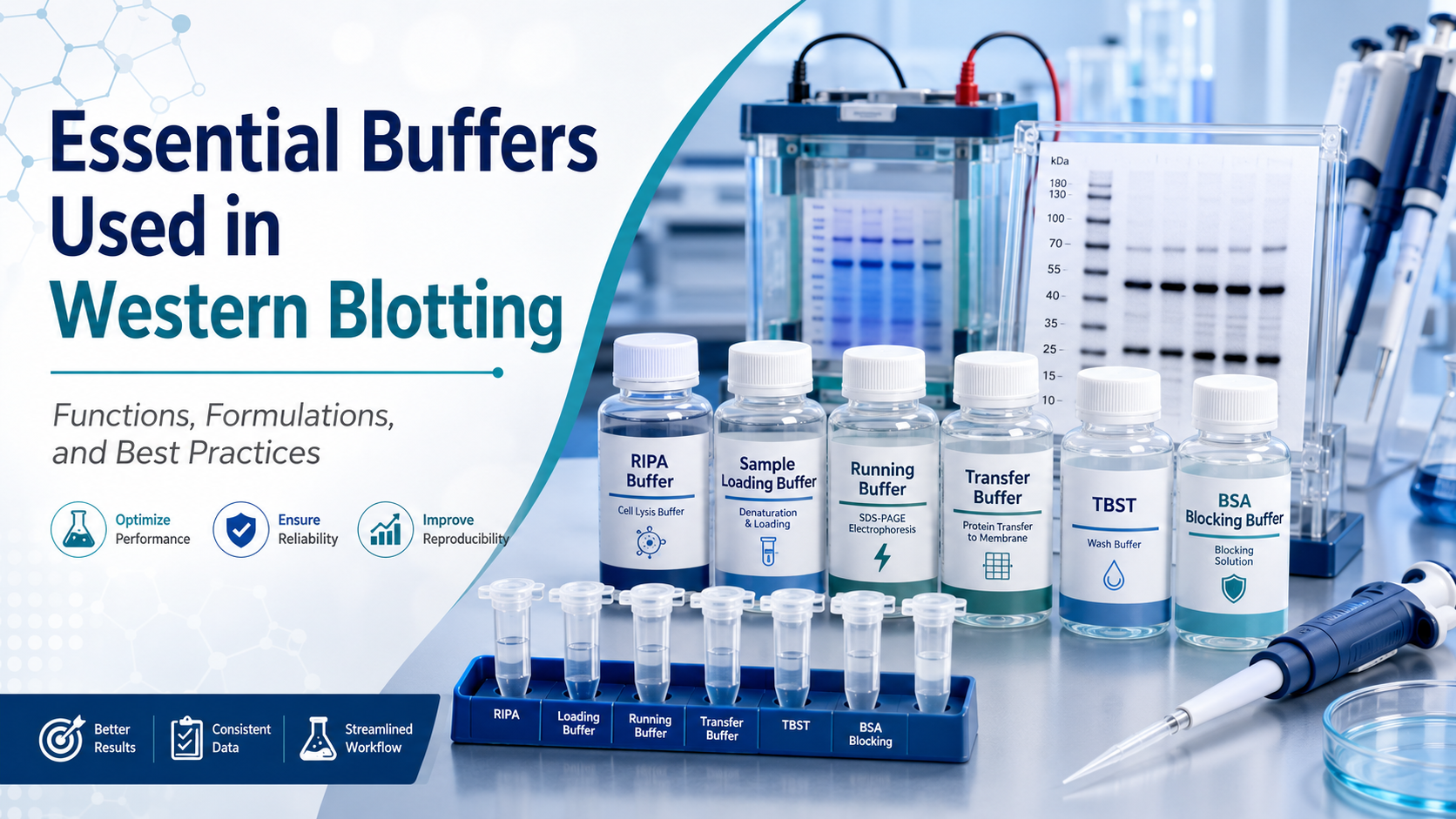
Western blotting, often abbreviated as WB, is one of the most widely used techniques in molecular biology, biochemistry, and biomedical research. It allows researchers to detect, identify, and semi-quantify specific proteins from complex biological samples. Although antibodies, membranes, and detection systems often receive the most attention, the success of a Western blot experiment depends heavily on another critical component: buffers.
Western blot buffers are used throughout the workflow, from protein extraction and sample preparation to electrophoresis, transfer, blocking, antibody incubation, and washing. Each buffer has a specific role in maintaining protein stability, ensuring efficient separation, reducing background signal, and improving reproducibility. Choosing the right buffer system can significantly affect band clarity, signal intensity, and overall experimental reliability.
This article introduces the main buffers used in Western blotting, explains their functions, and discusses practical considerations for optimizing WB results.
1. Lysis Buffer: Extracting Proteins from Cells and Tissues
The Western blot workflow begins with protein extraction. A lysis buffer is used to disrupt cells or tissues and release proteins into solution. At the same time, it helps maintain protein stability and reduce degradation.
One of the most commonly used lysis buffers is RIPA buffer. RIPA buffer is a strong lysis buffer suitable for extracting total proteins from cells and tissues, including cytoplasmic, membrane-associated, and nuclear proteins. It usually contains Tris-HCl, sodium chloride, nonionic detergents such as NP-40 or Triton X-100, ionic detergents such as SDS, and sodium deoxycholate. Because of its strong detergent composition, RIPA buffer can efficiently solubilize proteins but may sometimes disrupt protein-protein interactions.
For more gentle extraction, NP-40 lysis buffer or Triton X-100-based lysis buffer may be used. These buffers are often preferred when researchers want to preserve protein complexes or extract soluble proteins under milder conditions.
Protease inhibitors are usually added freshly before use to prevent protein degradation. If the experiment involves phosphorylation analysis, phosphatase inhibitors should also be included. Keeping samples on ice during lysis is also important for maintaining protein integrity.
2. Sample Loading Buffer: Preparing Proteins for SDS-PAGE
After protein extraction and quantification, samples are mixed with sample loading buffer before electrophoresis. This buffer is also known as Laemmli sample buffer or SDS sample buffer.
A typical sample loading buffer contains SDS, glycerol, Tris-HCl, bromophenol blue, and a reducing agent such as β-mercaptoethanol or dithiothreitol. SDS denatures proteins and gives them a uniform negative charge, allowing separation mainly according to molecular weight. The reducing agent breaks disulfide bonds, helping proteins unfold more completely. Glycerol increases sample density so that samples sink into the wells, while bromophenol blue serves as a tracking dye during electrophoresis.
Before loading, samples are usually heated at 95°C for 5 minutes or at 70°C for 10 minutes, depending on the target protein. Some membrane proteins or aggregation-prone proteins may require milder heating to avoid precipitation or abnormal migration.
3. Running Buffer: Supporting Protein Separation During Electrophoresis
SDS-PAGE electrophoresis requires a running buffer, also called electrophoresis buffer. The most common system is Tris-Glycine-SDS running buffer.
This buffer typically contains Tris base, glycine, and SDS. Tris helps maintain pH, glycine participates in the ion front during electrophoresis, and SDS keeps proteins denatured and negatively charged. Together, these components allow proteins to migrate through the polyacrylamide gel according to size.
Running buffer quality directly affects band shape and separation efficiency. Old or incorrectly prepared running buffer may cause distorted bands, poor resolution, or uneven migration. Researchers should ensure that the buffer is freshly prepared or properly stored, and that the correct concentration is used. In many laboratories, 10× stock running buffer is prepared and diluted to 1× before use.
4. Gel Buffers: Forming Stacking and Separating Gels
In traditional SDS-PAGE gel preparation, two different gel buffers are commonly used: stacking gel buffer and separating gel buffer.
The stacking gel buffer usually has a lower pH, often around pH 6.8, while the separating gel buffer has a higher pH, commonly around pH 8.8. This pH difference helps concentrate proteins into a narrow band before they enter the separating gel, improving resolution.
The stacking gel aligns proteins into a sharp front, while the separating gel resolves proteins based on molecular weight. The percentage of acrylamide in the separating gel should be selected according to the size of the target protein. Lower percentage gels are better for large proteins, while higher percentage gels provide better separation of small proteins.
Although many researchers now use precast gels, understanding the role of gel buffers remains important for troubleshooting Western blot problems.
5. Transfer Buffer: Moving Proteins from Gel to Membrane
After electrophoresis, proteins must be transferred from the gel onto a membrane, usually PVDF or nitrocellulose. This step requires transfer buffer.
A standard wet transfer buffer often contains Tris, glycine, methanol, and water. Methanol helps remove SDS from proteins and improves protein binding to the membrane. It also helps stabilize the gel during transfer. However, excessive methanol may reduce the transfer efficiency of large proteins because it can cause gel shrinkage.
For large proteins, researchers may reduce methanol concentration or add a small amount of SDS to improve transfer efficiency. For small proteins, methanol can help prevent proteins from passing through the membrane. Therefore, transfer buffer composition should be adjusted based on target protein size, membrane type, and transfer method.
PVDF membranes must be activated with methanol before use, while nitrocellulose membranes are usually equilibrated directly in transfer buffer. Proper buffer preparation and membrane handling are essential for efficient and uniform protein transfer.
6. Blocking Buffer: Reducing Non-Specific Binding
After transfer, the membrane contains many unoccupied binding sites. If these sites are not blocked, antibodies may bind non-specifically, causing high background signal. Blocking buffer is used to reduce this non-specific binding.
Common blocking buffers include 5% non-fat dry milk in TBST or PBST, and 5% BSA in TBST or PBST. Non-fat milk is economical and effective for many routine Western blot experiments. However, milk contains casein and endogenous phosphoproteins, which may interfere with phospho-specific antibodies. For detecting phosphorylated proteins, BSA is usually preferred.
The choice between milk and BSA depends on the antibody and target protein. Some antibodies perform better in milk, while others require BSA to reduce background or improve signal specificity. Blocking is commonly performed for 1 hour at room temperature or overnight at 4°C.
7. Washing Buffer: Removing Unbound Antibodies
Washing steps are essential for reducing background and improving signal-to-noise ratio. The most commonly used washing buffers are TBST and PBST.
TBST consists of Tris-buffered saline with Tween-20, while PBST consists of phosphate-buffered saline with Tween-20. Tween-20 is a mild detergent that helps remove loosely bound antibodies from the membrane. A typical concentration is 0.05% to 0.1%, although this may be adjusted depending on background level and antibody performance.

Washing is usually performed after primary antibody incubation and again after secondary antibody incubation. A common washing protocol is three washes of 5 to 10 minutes each. Insufficient washing can lead to high background, while overly harsh washing may reduce weak but specific signals.
TBST is widely used in Western blotting, but PBST may be suitable for some antibody systems. However, phosphate-containing buffers may not be ideal for certain phosphoprotein detection workflows or alkaline phosphatase-based detection systems.
8. Antibody Dilution Buffer: Maintaining Antibody Specificity
Primary and secondary antibodies are diluted in antibody dilution buffer before incubation with the membrane. This buffer is often based on TBST or PBST and may contain blocking agents such as BSA or non-fat milk.
The antibody dilution buffer helps stabilize antibodies and minimize non-specific binding during incubation. Primary antibody dilution conditions are especially important because they strongly influence signal specificity. Overly concentrated antibodies may produce high background or non-specific bands, while overly diluted antibodies may generate weak signals.
For many targets, primary antibodies are incubated overnight at 4°C to improve binding specificity. Secondary antibodies are commonly incubated for 1 hour at room temperature. The optimal dilution ratio should follow the antibody supplier’s recommendation and be adjusted experimentally if needed.
9. PBS and TBS: Basic Buffer Systems in Western Blotting
PBS and TBS are two foundational buffer systems used in Western blot workflows.
PBS, or phosphate-buffered saline, contains phosphate salts and sodium chloride. It provides physiological ionic strength and stable pH. TBS, or Tris-buffered saline, contains Tris and sodium chloride. TBS is commonly used in Western blotting because it is compatible with many antibody and detection systems.
The choice between PBS and TBS may depend on the antibody, target protein, and detection chemistry. In many chemiluminescent Western blot protocols, TBST is the standard washing and antibody dilution buffer. However, some antibodies may produce better results with PBST.
10. Practical Tips for Western Blot Buffer Optimization
Western blotting is sensitive to buffer composition, pH, detergent concentration, and storage conditions. To improve reproducibility, researchers should prepare buffers accurately, use high-quality reagents, and label stock solutions clearly.
Fresh inhibitors should be added to lysis buffer immediately before use. Running buffer and transfer buffer should be prepared at the correct dilution. Blocking and antibody dilution buffers should be selected according to the target and antibody type. For phosphoproteins, BSA-based blocking and dilution buffers are usually safer than milk-based buffers.
If high background occurs, increasing wash time, optimizing antibody dilution, or changing the blocking buffer may help. If the target signal is weak, researchers may optimize protein loading amount, transfer conditions, antibody concentration, or incubation time.
Conclusion
Buffers are essential components of every Western blot experiment. From lysis buffer and sample loading buffer to running buffer, transfer buffer, blocking buffer, washing buffer, and antibody dilution buffer, each solution plays a specific role in protein extraction, separation, transfer, detection, and signal optimization.
A successful Western blot does not depend only on antibodies or detection reagents. It also depends on carefully selected and properly prepared buffers. Understanding how each buffer works allows researchers to troubleshoot problems more effectively and obtain clearer, more reliable protein detection results.
For laboratories performing routine protein analysis, optimizing Western blot buffers is one of the most practical ways to improve experimental consistency, reduce background, and generate publication-quality results.
FireGene, light your research with passion, innovation, and profession.





