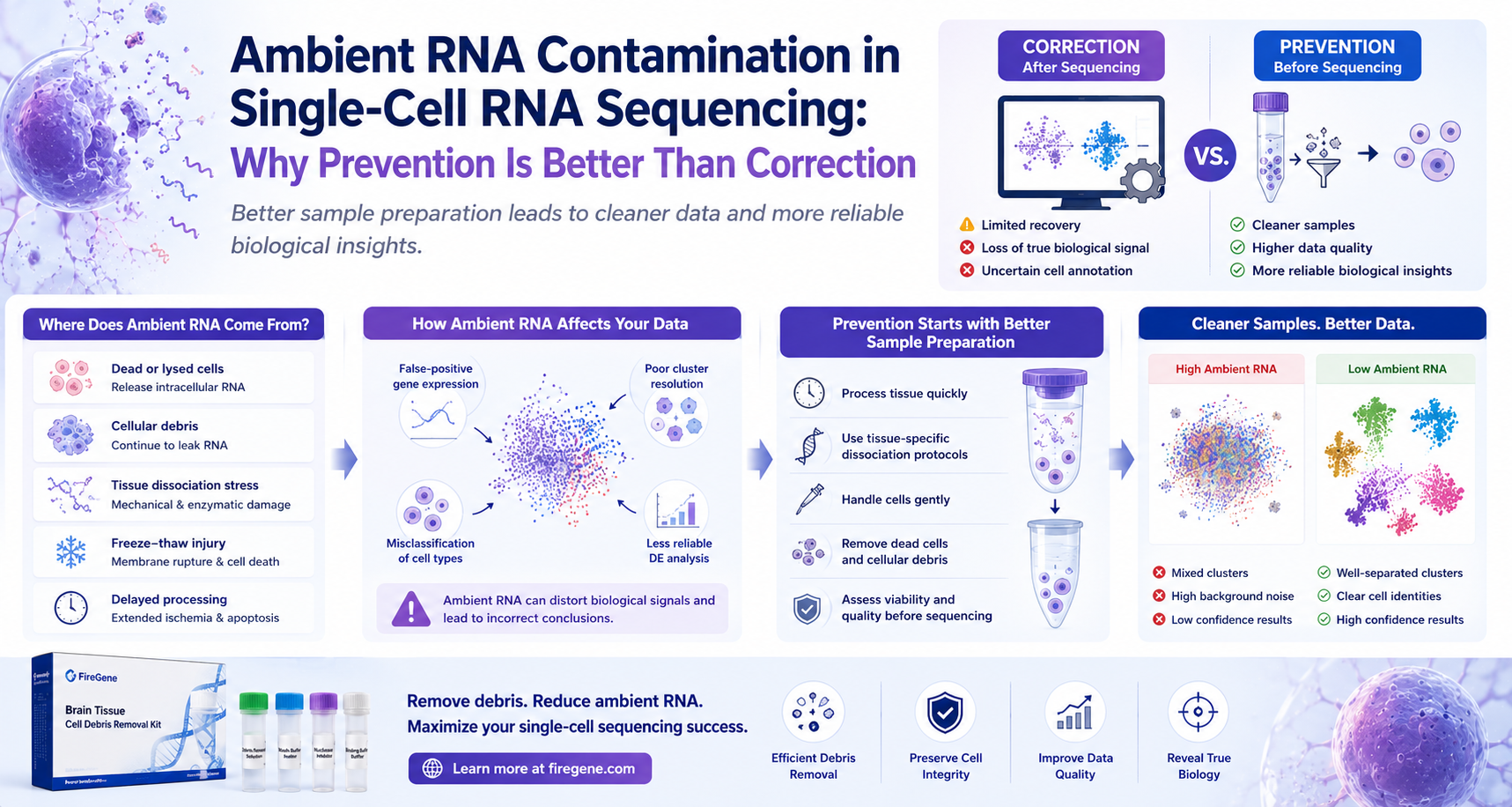
How Better Sample Preparation Leads to Cleaner Data and More Reliable Biological Discoveries
Single-cell RNA sequencing (scRNA-seq) has transformed biological research by enabling scientists to characterize complex tissues one cell at a time. From mapping cellular diversity in the Human Cell Atlas to identifying rare immune populations in cancer and uncovering neuronal subtypes in neurodegenerative disease, droplet-based single-cell technologies have become indispensable across modern life science research.
Yet despite remarkable advances in sequencing chemistry and computational biology, one persistent technical challenge continues to affect data quality in laboratories around the world:
Ambient RNA contamination.
Unlike sequencing errors or batch effects, ambient RNA originates long before a sample reaches the sequencer. It is generated during tissue dissociation and sample preparation when damaged or lysed cells release intracellular RNA into the surrounding suspension. These free RNA molecules can subsequently be encapsulated into droplets together with intact cells, introducing transcriptional signals that do not truly belong to those cells.
The consequences can be substantial.
Ambient RNA contamination may lead to:
- False-positive gene expression
- Distorted cell identities
- Poor clustering resolution
- Reduced differential expression accuracy
- Artificial cell-state transitions
- Misinterpretation of biological pathways
Over the past several years, computational tools such as SoupX, CellBender, DecontX, and related algorithms have become widely adopted to estimate and remove ambient RNA after sequencing.
However, these tools address only part of the problem.
They cannot recover biological information that has already been lost, nor can they fully eliminate contamination introduced during sample preparation.
Increasingly, experienced single-cell laboratories recognize a simple but important principle:
The best way to reduce ambient RNA is to prevent it from being generated in the first place.
This article explores the biological origin of ambient RNA, why it has become one of the most significant technical challenges in droplet-based sequencing, and how optimized sample preparation—including effective removal of damaged cells and cellular debris—can dramatically improve sequencing quality before a single sequencing read is generated.
Why Ambient RNA Has Become One of the Biggest Challenges in scRNA-seq
The earliest generations of single-cell RNA sequencing focused primarily on maximizing cell recovery and sequencing throughput.
As technologies matured, sequencing depth increased dramatically, allowing researchers to detect thousands of genes from tens of thousands—or even millions—of individual cells in a single experiment.
Ironically, this remarkable increase in sensitivity also made sequencing platforms more susceptible to contamination.
Modern droplet-based systems are capable of capturing extremely small amounts of RNA.
While this enables the detection of low-abundance transcripts, it also means that extracellular RNA released from damaged cells is more likely to be incorporated into sequencing libraries.
As a result, technical artifacts that were once relatively minor have become increasingly important determinants of data quality.
Large-scale benchmarking studies have consistently identified ambient RNA contamination as one of the major sources of technical variation in droplet-based scRNA-seq datasets, particularly when working with fragile tissues such as adult brain, inflamed organs, solid tumors, and cryopreserved clinical specimens.
For researchers investing significant time and resources into single-cell studies, minimizing ambient RNA is no longer simply a matter of computational refinement—it is an essential component of experimental design.
What Is Ambient RNA?
Ambient RNA refers to extracellular RNA molecules that are present in the cell suspension but are no longer contained within intact, viable cells.
Unlike the messenger RNA naturally found inside healthy cells, ambient RNA exists freely in the surrounding solution after being released from damaged or lysed cells.
During droplet generation, these free RNA molecules may enter droplets independently or together with intact cells.
Consequently, sequencing instruments cannot distinguish between:
- RNA originating from the target cell, and
- RNA originating from the surrounding environment.
The resulting sequencing library therefore contains a mixture of genuine biological signals and contaminating transcripts.
This contamination is particularly problematic because it often involves highly expressed genes released from abundant cell types, creating the appearance that unrelated cell populations express markers they do not biologically possess.
For example, hemoglobin transcripts released from lysed red blood cells may appear in immune cells, while neuronal transcripts from damaged neurons may be detected in glial populations.
Without careful quality control, these artifacts can lead to incorrect biological conclusions.
Where Does Ambient RNA Come From?
One of the most common misconceptions in single-cell sequencing is that ambient RNA is an unavoidable consequence of droplet-based technology.
In reality, ambient RNA is primarily generated during sample preparation—not during sequencing itself.
Before a cell suspension is ever loaded onto a sequencing platform, tissues undergo multiple processing steps, each of which has the potential to damage cells and release intracellular RNA.
The primary sources of ambient RNA include:
- Mechanical tissue disruption
- Enzymatic dissociation
- Cell apoptosis
- Necrotic tissue
- Freeze-thaw injury
- Prolonged sample handling
- Excessive centrifugation
- Poor storage conditions
Each damaged cell becomes a potential source of extracellular RNA.
As the proportion of damaged cells increases, ambient RNA accumulates within the suspension, creating an increasingly challenging environment for downstream sequencing.
Importantly, even a relatively small percentage of lysed cells can release a surprisingly large amount of RNA because intracellular mRNA concentrations are substantially higher than those found in the extracellular environment.
For this reason, preventing cellular damage during sample preparation has become one of the most effective strategies for minimizing ambient RNA contamination.
How Tissue Dissociation Generates Ambient RNA
Tissue dissociation is arguably the most critical—and most stressful—step in every single-cell RNA sequencing workflow.
Its objective is straightforward:
Convert intact tissue into a high-quality suspension of individual viable cells.
However, every dissociation protocol represents a compromise.
Researchers must digest extracellular matrix sufficiently to release cells while simultaneously preserving membrane integrity.
Achieving both goals perfectly is rarely possible.
Several mechanisms contribute to RNA release during dissociation.
Mechanical Damage
Mechanical disruption remains one of the largest contributors to ambient RNA generation.
Examples include:
- Excessive trituration
- Narrow pipette tips
- Vigorous vortexing
- High-speed centrifugation
- Repeated resuspension
Although these procedures improve tissue dissociation, they also increase the likelihood of membrane rupture.
Fragile cells—including mature neurons, hepatocytes, endothelial cells, and certain stem cell populations—are particularly susceptible.
Once membrane integrity is compromised, cytoplasmic RNA rapidly leaks into the surrounding suspension.
Enzymatic Overdigestion
Enzymes are indispensable for releasing individual cells from intact tissue.
However, prolonged enzymatic exposure may weaken plasma membranes and accelerate apoptosis.
Researchers often assume that longer digestion produces higher yields.
In reality, excessive digestion frequently produces:
- More dead cells
- More debris
- More extracellular RNA
- Lower overall sequencing quality
Optimizing digestion time is therefore essential for balancing cell recovery with preservation of RNA integrity.
Delayed Tissue Processing
Time is one of the most underestimated variables in single-cell sequencing.
Following tissue collection, gene expression profiles begin changing almost immediately.
As ischemia progresses:
- Cells experience metabolic stress.
- Membrane integrity declines.
- Apoptosis increases.
- RNA degradation accelerates.
Every additional hour before processing increases the probability of ambient RNA contamination.
Consequently, many sequencing core facilities strongly recommend minimizing the interval between tissue collection and dissociation whenever possible.
Freeze–Thaw Injury
Cryopreserved samples have become increasingly valuable in translational research.
Biobanks now store enormous collections of frozen tissues for retrospective studies and multicenter collaborations.
Despite these advantages, freeze-thaw cycles inevitably damage a proportion of cells.
Ice crystal formation disrupts plasma membranes, resulting in:
- Cell lysis
- Apoptosis
- Extracellular DNA release
- Increased ambient RNA
This explains why frozen clinical specimens often require additional cleanup before sequencing.
Which Tissues Produce the Highest Levels of Ambient RNA?
Although ambient RNA can occur in virtually any sample, certain tissues consistently present greater challenges.
Brain Tissue
Brain tissue is widely recognized as one of the most difficult tissues for scRNA-seq.
Several characteristics contribute to elevated ambient RNA:
- Fragile neuronal membranes
- Long axons and dendrites
- Extensive myelin
- High lipid content
- Disease-associated degeneration
Adult brain tissue generally produces substantially more ambient RNA than embryonic brain because mature neurons are considerably more susceptible to mechanical damage.
Neurodegenerative disease models—including Alzheimer's disease, Parkinson's disease, Huntington's disease, and amyotrophic lateral sclerosis (ALS)—often contain extensive neuronal loss before tissue processing even begins.
Solid Tumors
Tumor tissues frequently contain:
- Necrotic regions
- Hypoxic microenvironments
- Dead immune cells
- Fibrotic stroma
- Damaged endothelial cells
These characteristics make tumors major sources of extracellular RNA.
Additionally, heterogeneous tumor architecture often requires prolonged tissue digestion, further increasing the likelihood of cellular damage.
Inflamed Organs
Inflammation is frequently accompanied by active cell death.
Diseases affecting:
- Liver
- Kidney
- Lung
- Intestine
often generate substantial amounts of apoptotic material during tissue processing.
Inflammatory infiltrates also contribute abundant extracellular nucleic acids that complicate downstream sequencing.
Archived Clinical Samples
Human biopsy specimens are increasingly valuable for translatform and translational research.
However, clinical workflows rarely allow immediate tissue processing.
Transportation delays, temporary storage, and cryopreservation collectively increase cellular stress before dissociation begins.
As a result, archived clinical samples frequently exhibit higher ambient RNA contamination than freshly processed experimental tissues.
Ambient RNA vs. Cell Debris vs. Dead Cells
These three terms are often used interchangeably, but they describe distinct components of sample quality.
| Feature | Dead Cells | Cell Debris | Ambient RNA |
|---|---|---|---|
| Biological structure | Intact but non-viable cells | Fragmented cellular material | Free RNA molecules |
| Visible microscopically | Yes | Usually | No |
| Source | Apoptosis or necrosis | Cell rupture | Released from damaged or lysed cells |
| Can be physically removed | Often | Yes | Mostly prevented rather than removed |
| Major consequence | Lower viability | Reduced sample purity | False gene expression signals |
Importantly, these components are biologically connected.
A typical progression is:
Healthy Cell → Dead Cell → Cell Rupture → Cell Debris + Ambient RNA
This cascade illustrates why preventing cell damage at the earliest possible stage has such a profound impact on downstream sequencing quality.
Reducing dead cells naturally reduces debris.
Reducing debris consequently reduces ambient RNA.
Rather than treating these issues independently, modern sample preparation workflows increasingly address them together through comprehensive quality control strategies.
Why Computational Correction Is Not Enough
In recent years, computational tools have become increasingly sophisticated.
Algorithms such as SoupX, CellBender, DecontX, and related methods estimate background RNA by modeling expression patterns across droplets.
These tools have greatly improved the interpretation of droplet-based datasets.
However, they should be viewed as damage control—not damage prevention.
Computational correction has several important limitations:
- It relies on statistical assumptions rather than direct biological measurements.
- It cannot restore transcripts lost from damaged cells.
- It cannot recover fragile cell populations that were destroyed during dissociation.
- It cannot reverse stress-induced transcriptional changes.
- It may inadvertently remove genuine low-abundance biological signals while correcting contamination.
An analogy is useful:
Computational correction is like removing dust from a digital photograph after it has been taken. Preventing ambient RNA is like cleaning the camera lens before taking the picture.
Both approaches improve the final image, but prevention preserves far more original information.
For this reason, leading sequencing laboratories increasingly emphasize prevention-first workflows, combining optimized tissue handling, gentle dissociation, rapid processing, debris removal, and viability assessment before a sample ever reaches the sequencing instrument.
This philosophy not only reduces ambient RNA but also improves the overall biological fidelity of single-cell datasets.
Best Practices to Prevent Ambient RNA Before Sequencing
Unlike sequencing artifacts that can only be addressed after data generation, ambient RNA contamination is largely preventable.
Although no protocol can completely eliminate extracellular RNA, careful optimization of sample preparation can dramatically reduce contamination before sequencing begins.
Increasingly, experienced sequencing facilities emphasize a prevention-first strategy built around several key principles.
1. Process Tissue as Quickly as Possible
The interval between tissue collection and dissociation has a direct impact on sample quality.
Following tissue excision, cells immediately begin responding to ischemia, nutrient deprivation, and mechanical stress.
As processing time increases:
- Cell viability decreases.
- Membrane integrity deteriorates.
- Apoptosis accelerates.
- More intracellular RNA leaks into the extracellular environment.
Whenever possible, tissues should be processed immediately after collection.
For clinical samples where immediate processing is not feasible, standardized preservation procedures become especially important.
2. Use Tissue-Specific Dissociation Protocols
Different tissues require different enzymatic and mechanical dissociation strategies.
Brain tissue, for example, contains highly fragile neurons and abundant myelin, whereas fibrotic liver or kidney tissue contains dense extracellular matrix that requires more aggressive digestion.
Applying a "one-size-fits-all" dissociation protocol often results in unnecessary cellular damage.
Instead, researchers should optimize:
- Enzyme composition
- Digestion duration
- Temperature
- Mechanical dissociation intensity
- Tissue fragment size
Tailoring protocols to individual tissue types helps maximize viable cell recovery while minimizing RNA release.
3. Handle Cell Suspensions Gently
Many cells remain extremely fragile after dissociation.
Excessive manipulation during downstream processing can significantly increase cell lysis.
Researchers should avoid:
- Vigorous pipetting
- Excessive vortexing
- Harsh centrifugation
- Repeated pellet resuspension
Instead, gentle handling throughout the workflow preserves membrane integrity and reduces the generation of extracellular RNA.
4. Remove Damaged Cells and Cellular Debris Before Library Preparation
One of the most effective strategies for limiting ambient RNA is reducing the source of contamination itself.
Damaged cells and cellular debris continue releasing RNA throughout sample preparation.
If these contaminants remain in the suspension until droplet generation, extracellular RNA becomes increasingly difficult to avoid.
Consequently, many laboratories now include dedicated cleanup procedures before final cell counting and library preparation.
Removing damaged cellular material not only improves suspension purity but also reduces the continuous release of RNA into the surrounding solution.
This preventative approach is considerably more effective than relying exclusively on computational correction after sequencing.
Why Cell Debris Removal Helps Reduce Ambient RNA
Although cell debris removal is often discussed in the context of improving sample cleanliness, its benefits extend much further.
Damaged cellular fragments represent ongoing sources of extracellular RNA.
As fragmented membranes continue to degrade, additional RNA is released into the suspension.
By removing these fragments before sequencing, researchers reduce one of the primary contributors to ambient RNA contamination.
This is particularly important when processing:
- Adult brain tissue
- Glioblastoma samples
- Alzheimer's disease models
- Parkinson's disease models
- Cryopreserved tissue
- Necrotic tumors
- Fibrotic organs
These tissues naturally generate higher levels of cellular damage during dissociation and therefore benefit substantially from optimized cleanup procedures.
Supporting Cleaner Single-Cell Workflows with FireGene
As single-cell sequencing continues to expand into increasingly challenging sample types, standardized sample preparation reagents are becoming an essential component of reproducible research workflows.
The FireGene Brain Tissue Cell Debris Removal Kit was developed to help researchers efficiently remove cellular debris from dissociated brain tissue prior to downstream applications.
Rather than replacing optimized dissociation protocols, the kit is designed to complement existing workflows by improving sample purity after tissue digestion.
Typical downstream applications include:
- Single-cell RNA sequencing (scRNA-seq)
- Single-nucleus RNA sequencing (snRNA-seq)
- Flow cytometry
- Fluorescence-activated cell sorting (FACS)
- Spatial transcriptomics
- Multiomic analyses
By reducing debris-rich contaminants before library preparation, researchers can generate cleaner cell suspensions that are better suited for high-quality sequencing experiments.
Learn more about the FireGene Brain Tissue Cell Debris Removal Kit:
https://firegene.com/products/brain-tissue-cell-debris-removal-kit?variant=47868495921364
Case Study: Prevention vs. Computational Correction
Consider two research groups studying immune cell heterogeneity within glioblastoma tissue.
Both laboratories use the same sequencing platform and generate comparable sequencing depth.
Laboratory A: Prevention-First Workflow
Before library preparation, the researchers perform:
- Optimized tissue dissociation
- Filtration
- Cell debris removal
- Viability assessment
- Cell concentration adjustment
Their dataset demonstrates:
- Low ambient RNA
- High library complexity
- Distinct immune cell clusters
- Robust differential expression
- Clear separation of macrophages, microglia, and T-cell populations
Laboratory B: Correction-First Workflow
The second laboratory performs tissue dissociation and immediately proceeds to sequencing, intending to remove contamination later using computational software.
Although SoupX and CellBender successfully reduce part of the background contamination, several issues remain:
- Weak expression of lineage-specific markers
- Reduced resolution of rare immune populations
- Increased uncertainty during cell annotation
- Loss of confidence in downstream pathway analyses
The difference between the two studies illustrates an increasingly accepted principle in single-cell research:
Computational correction can improve contaminated datasets, but it cannot fully recover biological information that was compromised during sample preparation.
High-quality sequencing begins with high-quality samples—not high-quality algorithms alone.
Sample Quality Checklist Before Sequencing
Before loading your sample onto a droplet-based sequencing platform, confirm the following:
✅ Tissue processed promptly after collection
✅ Tissue-specific dissociation protocol optimized
✅ Gentle mechanical handling maintained throughout processing
✅ Cell viability assessed
✅ Cell aggregates minimized
✅ Cellular debris removed
✅ Cell concentration accurately measured
✅ Sample filtered appropriately
✅ Suspension kept cold where appropriate
✅ Final suspension visually free of excessive debris
Following this checklist can substantially reduce ambient RNA generation while improving sequencing reproducibility across experiments and operators.
Frequently Asked Questions (FAQ)
1. What is ambient RNA contamination in single-cell RNA sequencing?
Ambient RNA refers to extracellular RNA molecules released from damaged or lysed cells during tissue processing. During droplet generation, these free RNA molecules may be captured together with viable cells, creating transcriptional signals that do not truly belong to the encapsulated cell.
Ambient RNA contamination is one of the most common technical artifacts in droplet-based scRNA-seq and can significantly affect downstream biological interpretation.
2. Why is ambient RNA more common in droplet-based sequencing?
Droplet-based platforms such as those used for high-throughput single-cell RNA sequencing are extremely sensitive and efficiently capture RNA molecules present within each droplet.
Unfortunately, sequencing instruments cannot distinguish between:
- RNA originating from intact cells
- RNA originating from the extracellular environment
Consequently, any RNA released during tissue dissociation may become incorporated into sequencing libraries.
3. Which tissues are most susceptible to ambient RNA contamination?
Although ambient RNA can occur in almost any tissue, contamination is particularly common in:
- Adult brain tissue
- Neurodegenerative disease models
- Solid tumors
- Necrotic tissues
- Fibrotic organs
- Cryopreserved clinical specimens
- Inflamed tissues
These samples often contain larger proportions of damaged or dying cells prior to sequencing.
4. Can ambient RNA affect cell clustering?
Yes.
Ambient RNA may introduce transcripts that artificially increase the similarity between unrelated cells.
This can result in:
- Poor cluster separation
- Incorrect cell annotation
- False-positive marker genes
- Misleading differential expression results
Reducing contamination before sequencing improves clustering accuracy and biological confidence.
5. Can computational tools completely eliminate ambient RNA?
No.
Software such as SoupX, CellBender, and DecontX provides valuable post-sequencing correction by estimating background contamination.
However, these algorithms cannot:
- Recover RNA lost from lysed cells
- Restore destroyed cell populations
- Reverse transcriptional stress responses
- Fully reconstruct the original biological state
Prevention remains considerably more effective than correction.
6. How can researchers reduce ambient RNA before sequencing?
Several practical strategies help minimize contamination:
- Rapid tissue processing
- Tissue-specific dissociation protocols
- Gentle mechanical handling
- Maintaining high cell viability
- Removing damaged cells and cellular debris
- Filtering aggregates
- Performing quality control before library preparation
Collectively, these measures reduce RNA release throughout sample preparation.
7. Why is cell debris removal important for ambient RNA prevention?
Cell debris represents a continuing source of extracellular RNA.
As damaged membranes degrade, additional RNA is released into the surrounding suspension.
Removing debris before sequencing reduces one of the primary sources of ambient RNA contamination while simultaneously improving sample purity.
For debris-rich tissues such as adult brain, this step has become an increasingly common component of optimized single-cell workflows.
Learn more about the FireGene Brain Tissue Cell Debris Removal Kit.
8. Does ambient RNA affect single-nucleus RNA sequencing?
Yes.
Although snRNA-seq isolates nuclei rather than whole cells, extracellular RNA released during tissue processing can still contaminate nuclei suspensions.
Careful tissue handling and optimized cleanup remain important for both scRNA-seq and snRNA-seq workflows.
Conclusion
As single-cell RNA sequencing continues to reshape biomedical research, expectations for data quality continue to rise.
Researchers are no longer satisfied with simply generating large datasets—they require datasets that accurately reflect the biology of the original tissue.
Ambient RNA contamination represents one of the most significant obstacles to achieving this goal.
Although computational correction has advanced considerably, it should be viewed as a complementary tool rather than a substitute for good laboratory practice.
The most effective strategy remains preventing ambient RNA before sequencing begins.
By minimizing cellular damage, optimizing tissue dissociation, preserving viability, and removing damaged cells and cellular debris, researchers can generate cleaner suspensions that support more reliable biological discovery.
For laboratories working with fragile or debris-rich tissues—including brain, tumors, cryopreserved specimens, and neurodegenerative disease models—implementing standardized cleanup procedures before library preparation can substantially improve sequencing consistency and reproducibility.
The FireGene Brain Tissue Cell Debris Removal Kit was developed to support this critical stage of the workflow by helping researchers generate cleaner cell suspensions for downstream applications including:
- Single-cell RNA sequencing (scRNA-seq)
- Single-nucleus RNA sequencing (snRNA-seq)
- Flow cytometry
- Fluorescence-activated cell sorting (FACS)
- Spatial transcriptomics
- Single-cell multiomics
Whether your goal is to map cellular heterogeneity, discover disease-associated cell states, or build next-generation cellular atlases, preventing ambient RNA contamination begins with optimizing sample preparation—not with correcting sequencing data afterward.
References
- Young MD, Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience. 2020;9(12):giaa151.
- Fleming SJ, Marioni JC, Babadi M. CellBender remove-background: deep generative modeling for background correction in single-cell RNA sequencing. Nature Methods. 2023.
- Yang S, Corbett SE, Koga Y, et al. DecontX: decontamination of ambient RNA in single-cell RNA-seq with Bayesian modeling. Genome Biology. 2020.
- Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Molecular Systems Biology. 2019;15:e8746.
- Stuart T, Satija R. Integrative single-cell analysis. Nature Reviews Genetics. 2019;20:257–272.
- Mereu E, et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nature Biotechnology. 2020;38:747–755.
- Vieth B, Parekh S, Ziegenhain C, et al. A systematic evaluation of single-cell RNA-sequencing analysis pipelines. Nature Communications. 2019;10:4667.
- McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Systems. 2019;8(4):329–337.
- Regev A, et al. The Human Cell Atlas. eLife. 2017;6:e27041.
FireGene Single-Cell Sample Prep
Looking for validated dissociation kits?
FireGene's tissue dissociation kits are optimized for specific organ types — brain, tumor, liver, GI, reproductive, and more. Validated for 10x Genomics Chromium and BD Rhapsody workflows.





