Frequently Asked Questions (FAQ)
1. What is cell debris in single-cell RNA sequencing?
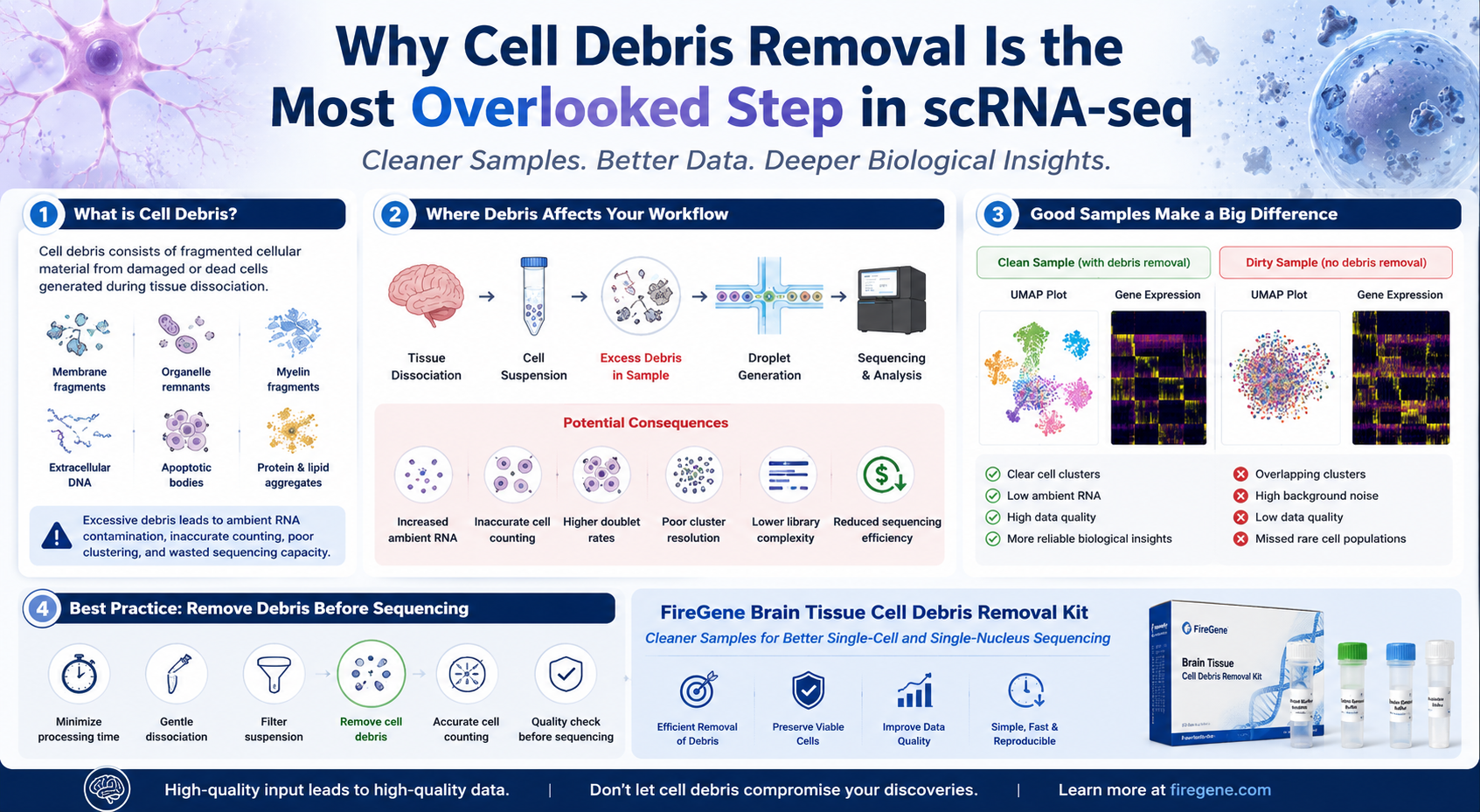
Cell debris refers to fragmented cellular material generated during tissue dissociation or from dead and damaged cells. It may include membrane fragments, organelles, extracellular DNA, myelin, apoptotic bodies, and other non-viable cellular components.
Although debris does not represent intact cells, it can significantly interfere with downstream workflows by increasing background contamination, complicating cell counting, and contributing to ambient RNA.
2. Why is cell debris a problem for scRNA-seq?
Excessive debris can negatively affect almost every stage of a single-cell sequencing experiment.
Potential consequences include:
- Reduced cell counting accuracy
- Increased ambient RNA contamination
- Lower library complexity
- Poor droplet encapsulation efficiency
- Higher doublet rates
- Reduced recovery of rare cell populations
- Less accurate cell clustering
Because many of these issues originate before sequencing, they are difficult—or impossible—to completely correct using computational methods alone.
3. Does every tissue require cell debris removal?
Not necessarily.
The amount of debris varies depending on tissue type, sample quality, and experimental workflow.
Debris removal is particularly beneficial for:
- Adult brain tissue
- Neurodegenerative disease models
- Glioblastoma and other brain tumors
- Cryopreserved tissue
- Inflamed tissues
- Fibrotic organs
- Necrotic tumor specimens
- Aged animal tissues
For these sample types, dedicated cleanup can substantially improve downstream sequencing performance.
4. What is the difference between cell debris and dead cells?
Although related, they are not identical.
Dead cells remain intact but are no longer viable.
Cell debris consists of fragmented cellular components produced after cells rupture or undergo extensive damage.
Dead cells frequently become a major source of debris and extracellular RNA during sample preparation.
5. Can computational software remove the effects of cell debris?
Only partially.
Several excellent computational tools—including SoupX, CellBender, and DecontX—can estimate background contamination after sequencing.
However, these algorithms cannot:
- Recover destroyed cells
- Restore lost cell populations
- Reverse processing-induced stress responses
- Eliminate all technical artifacts
Preventing contamination during sample preparation remains the preferred strategy.
6. When should cell debris removal be performed?
For most workflows, debris removal is recommended after tissue dissociation and filtration but before final cell counting and library preparation.
This sequence allows researchers to:
- Remove damaged cellular material
- Improve cell counting accuracy
- Obtain more reliable viability measurements
- Generate cleaner suspensions for downstream sequencing
7. Does debris removal reduce cell recovery?
A properly optimized debris removal protocol is designed to remove unwanted contaminants while preserving viable cells.
The objective is not to maximize the total number of particles recovered, but to maximize the recovery of biologically meaningful, intact cells suitable for downstream analysis.
8. Is cell debris removal important for single-nucleus RNA sequencing?
Yes.
Although snRNA-seq isolates nuclei rather than whole cells, damaged tissue can still contain abundant cellular fragments, extracellular DNA, and other contaminants that interfere with nuclei purification.
For frozen brain tissue and archived clinical specimens, debris removal may improve nuclei purity and downstream data quality.
9. Which downstream applications benefit from cleaner cell suspensions?
High-quality debris removal can support numerous downstream applications, including:
- Single-cell RNA sequencing (scRNA-seq)
- Single-nucleus RNA sequencing (snRNA-seq)
- Flow cytometry
- Fluorescence-activated cell sorting (FACS)
- Spatial transcriptomics
- CITE-seq
- Multiome ATAC + Gene Expression
- Cell culture following tissue dissociation
10. How can I improve sample quality before sequencing?
Researchers should establish a standardized workflow that includes:
- Rapid tissue collection and processing
- Tissue-specific dissociation protocols
- Gentle mechanical handling
- Effective filtration
- Cell debris removal
- Cell viability assessment
- Aggregate inspection
- Accurate cell counting
- Optimized loading concentration
Consistent implementation of these steps improves reproducibility and helps ensure that sequencing data accurately reflect the original biology.
Conclusion
Single-cell RNA sequencing has transformed our ability to explore cellular diversity, but the quality of every dataset still depends on one fundamental principle:
High-quality sequencing begins with high-quality sample preparation.
While advances in sequencing chemistry and computational biology continue to expand what is technically possible, they cannot compensate for poor biological input.
Cell debris is more than an inconvenience observed under the microscope. It represents a significant source of technical variability that can reduce sequencing efficiency, increase ambient RNA contamination, compromise clustering accuracy, and ultimately obscure meaningful biological discoveries.
As single-cell technologies continue to evolve toward larger cell atlas projects, spatial transcriptomics, and integrated multiomics, the importance of standardized sample preparation will only continue to grow.
Increasingly, leading research laboratories view cell debris removal as an essential quality control step rather than an optional refinement.
By combining optimized tissue dissociation, gentle sample handling, effective filtration, rigorous quality assessment, and dedicated debris removal, researchers can generate cleaner suspensions that support more reproducible and biologically meaningful results.
For laboratories working with brain tissue and other debris-rich samples, the FireGene Brain Tissue Cell Debris Removal Kit provides a standardized solution for improving sample purity before downstream applications such as:
- Single-cell RNA sequencing (scRNA-seq)
- Single-nucleus RNA sequencing (snRNA-seq)
- Flow cytometry
- Cell sorting
- Spatial transcriptomics
- Multiomics research
Whether your goal is to identify rare neuronal subtypes, investigate tumor heterogeneity, build a cellular atlas, or explore disease mechanisms at single-cell resolution, investing in sample quality before sequencing is one of the most effective ways to improve experimental success.
References
- Stuart T, Satija R. Integrative single-cell analysis. Nature Reviews Genetics. 2019;20:257–272.
- Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Molecular Systems Biology. 2019;15:e8746.
- Mereu E, et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nature Biotechnology. 2020;38:747–755.
- Vieth B, Parekh S, Ziegenhain C, et al. A systematic evaluation of single-cell RNA-sequencing analysis pipelines. Nature Communications. 2019;10:4667.
- Young MD, Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience. 2020;9(12):giaa151.
- McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Systems. 2019;8(4):329–337.
- Fleming SJ, Marioni JC, Babadi M. CellBender remove-background: deep generative modeling for single-cell RNA sequencing background correction. Nature Methods. 2023.
- Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biology. 2019;20:296.
- Regev A, et al. The Human Cell Atlas. eLife. 2017;6:e27041.
FireGene Single-Cell Sample Prep
Looking for validated dissociation kits?
FireGene's tissue dissociation kits are optimized for specific organ types — brain, tumor, liver, GI, reproductive, and more. Validated for 10x Genomics Chromium and BD Rhapsody workflows.





