The pharmaceutical industry is experiencing one of the most significant changes in endotoxin testing since the introduction of the Limulus Amebocyte Lysate (LAL) assay more than fifty years ago.
For decades, USP <85> Bacterial Endotoxins Test (BET) has been the global standard for detecting bacterial endotoxins in injectable drugs, biologics, vaccines, and medical devices. However, growing interest in sustainability, supply chain resilience, and recombinant biotechnology has driven the development of alternative testing approaches, culminating in the introduction of USP <86> Bacterial Endotoxins Test Using Recombinant Reagents.

Today, quality control laboratories face important questions:
- Should we continue using traditional TAL/LAL methods?
- Is Recombinant Factor C (rFC) accepted by regulators?
- What validation studies are required under USP <86>?
- Will recombinant methods eventually replace conventional endotoxin testing?
- How should validation teams prepare for future regulatory expectations?
This article provides a practical comparison of USP <85> and USP <86>, discusses real-world validation challenges, and outlines how pharmaceutical manufacturers can build a future-proof endotoxin testing strategy.
Why Endotoxin Testing Remains Critical
Bacterial endotoxins are lipopolysaccharides (LPS) originating from the outer membrane of Gram-negative bacteria.
Even extremely low levels of endotoxin contamination can trigger severe physiological responses, including:
- Fever
- Inflammation
- Hypotension
- Septic shock
- Organ dysfunction
Because injectable pharmaceuticals bypass the body's natural defense barriers, endotoxin control is considered one of the most important aspects of pharmaceutical quality assurance.
Failure to control endotoxin contamination can result in:
- Product recalls
- Regulatory observations
- Manufacturing delays
- Patient safety risks
As a result, endotoxin testing remains a core requirement throughout pharmaceutical development, manufacturing, validation, and batch release.
The Evolution of Endotoxin Testing
The evolution of endotoxin detection reflects broader changes in pharmaceutical quality control.
Traditional Era
- Rabbit Pyrogen Test (RPT)
- Animal-based pyrogen detection
Modern Compendial Era
- LAL assays
- TAL assays
- USP <85> standardization
Emerging Era
- Recombinant Factor C (rFC)
- Recombinant Cascade Reagents (rCR)
- USP <86>
Rather than replacing existing technologies overnight, the industry is gradually moving toward a hybrid analytical ecosystem where multiple validated methods coexist.

Understanding USP <85>: The Foundation of Modern Endotoxin Testing
USP <85> has served as the regulatory backbone of endotoxin testing for decades.
The chapter describes endotoxin detection methods based on lysate reagents derived from horseshoe crab blood.
These include:
Gel-Clot Method
The original compendial approach.
Advantages
- Simple implementation
- Low equipment requirements
- Global regulatory acceptance
Limitations
- Semi-quantitative
- Lower throughput
Kinetic Chromogenic Method
Measures color development generated by endotoxin activation.
Advantages
- High sensitivity
- Quantitative results
- Suitable for automation
Applications
- Biologics
- Injectable pharmaceuticals
- Process validation
Kinetic Turbidimetric Method
Measures increases in turbidity during the endotoxin reaction cascade.
Advantages
- Quantitative
- Broad dynamic range
- Widely accepted
Challenges Associated with Traditional LAL and TAL Testing
Although USP <85> remains highly reliable, several industry concerns have emerged.
1. Sustainability Concerns
Traditional lysate reagents are derived from horseshoe crabs.
As pharmaceutical testing demand continues to increase, environmental and sustainability considerations have received greater attention worldwide.
2. Supply Chain Risks
Manufacturers increasingly seek to reduce dependency on biological raw materials.
Potential concerns include:
- Limited harvesting regions
- Production variability
- Global supply disruptions
3. Regulatory Modernization
Regulatory agencies continue encouraging scientifically justified alternative methods when equivalent or superior performance can be demonstrated.
This trend ultimately led to the development of USP <86>.
What Is USP <86>?
USP <86> introduces bacterial endotoxin testing methods based on recombinant biotechnology.
Unlike traditional LAL or TAL reagents, recombinant methods use laboratory-produced proteins to detect endotoxin contamination.
The two most discussed technologies are:
Recombinant Factor C (rFC)
Factor C is the primary endotoxin-sensitive protein within the natural horseshoe crab coagulation cascade.
Scientists have successfully cloned and expressed this protein using recombinant technology.
When endotoxin is present, recombinant Factor C becomes activated and generates a measurable signal.
Recombinant Cascade Reagents (rCR)
These systems incorporate multiple recombinant proteins that replicate larger portions of the natural coagulation cascade.
The result is an entirely animal-free endotoxin detection platform.
USP <85> vs USP <86>: Key Differences
| Feature | USP <85> | USP <86> |
|---|---|---|
| Reagent Source | TAL/LAL | Recombinant proteins |
| Animal-Derived | Yes | No |
| Regulatory History | Established for decades | Newly introduced compendial chapter |
| Testing Platforms | Gel clot, chromogenic, turbidimetric | Recombinant fluorescence assays |
| Sustainability Profile | Moderate | Excellent |
| Validation Complexity | Well-established | Higher during implementation |
| Global Adoption | Extensive | Rapidly expanding |
| Supply Chain Dependence | Biological sourcing | Biotechnology manufacturing |
The Most Important Regulatory Reality: USP <86> Does Not Replace USP <85>
A common misconception is that USP <86> replaces USP <85>.
This is incorrect.
USP <85> remains the primary compendial standard for bacterial endotoxin testing.
USP <86> provides an additional validated pathway that organizations may adopt when appropriate scientific justification exists.
For most pharmaceutical manufacturers, the foreseeable future will involve:
- USP <85> for routine QC testing
- USP <86> for selected products and advanced applications
This dual-method environment is likely to remain the industry standard for years to come.

What Validation Teams Are Actually Seeing in Practice
One area often overlooked in discussions about USP <86> is implementation complexity.
Based on industry experience, the greatest challenge is not purchasing recombinant reagents—it is demonstrating comparability to existing validated methods.
Validation teams frequently encounter:
Matrix Interference
Complex formulations can behave differently under recombinant assays.
Examples include:
- Monoclonal antibodies
- Lipid nanoparticles
- Gene therapy vectors
- Cell therapy products
Bridging Studies
Organizations often need to demonstrate that results generated under USP <86> are equivalent to previously established USP <85> methods.
Documentation Burden
Regulatory submissions may require:
- Method comparison reports
- Validation protocols
- Risk assessments
- Scientific justification packages
These activities often represent the most time-consuming portion of implementation.
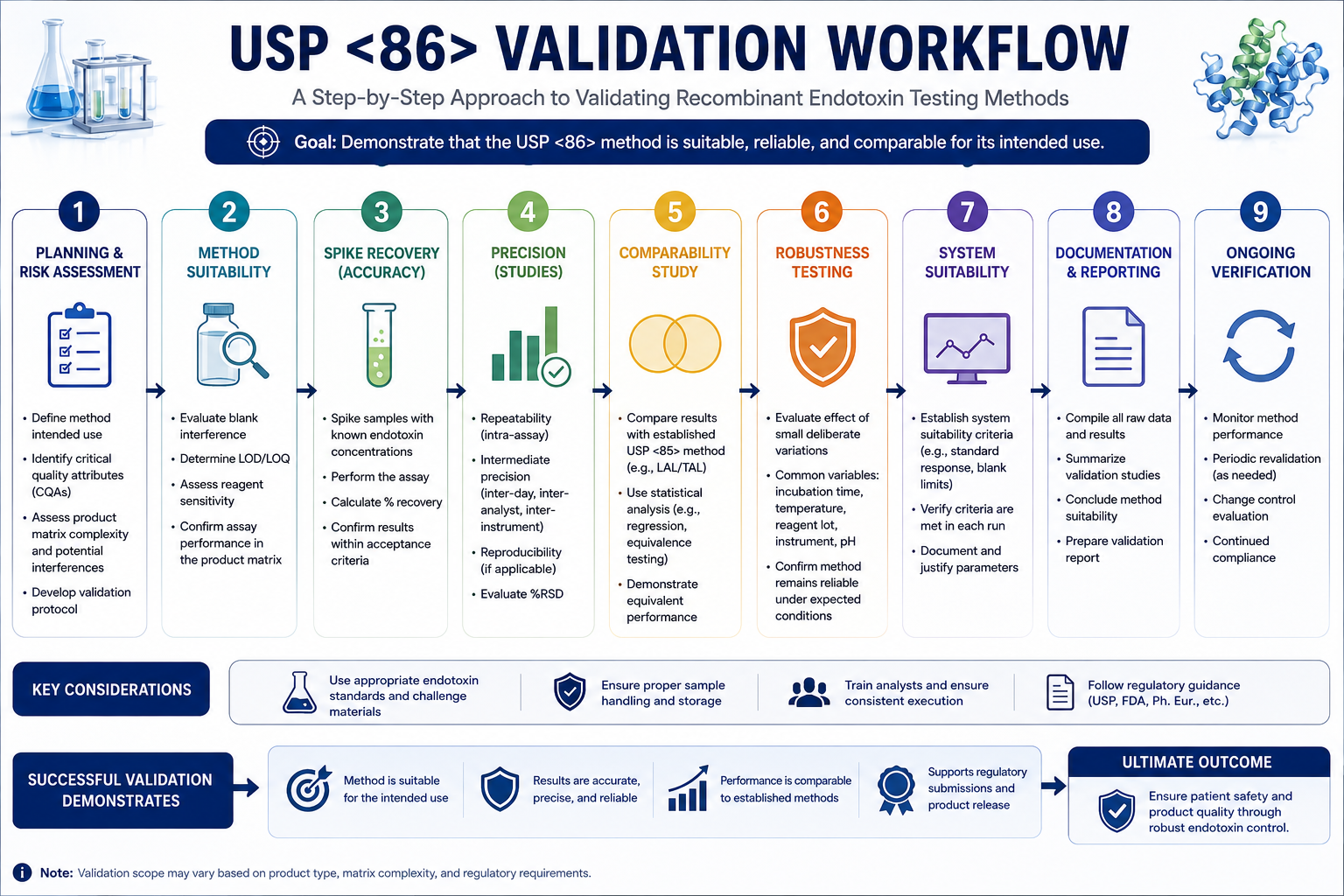
Validation Requirements Under USP <86>
Successful implementation requires a structured validation strategy.
Method Suitability Testing
Demonstrates that the assay accurately detects endotoxin within a specific product matrix.
Potential interferences may include:
- Proteins
- Surfactants
- Salts
- Preservatives
Spike Recovery Studies
Known endotoxin concentrations are added to product samples to confirm accurate recovery.
These studies help establish:
- Accuracy
- Matrix compatibility
- Detection reliability
Precision Assessment
Evaluates:
- Repeatability
- Intermediate precision
- Analyst-to-analyst variation
Robustness Studies
Common parameters include:
- Incubation temperature
- Timing variations
- Instrument differences
- Reagent lots
Method Comparability
One of the most important activities when transitioning from USP <85> to USP <86>.
The goal is to demonstrate equivalent performance between both approaches.
Why Endotoxin Challenge Materials Matter More Than Ever
As validation programs become increasingly sophisticated, standardized endotoxin reference materials play a critical role.
Many pharmaceutical manufacturers use Endotoxin Challenge Vials (ECVs) during:
- Depyrogenation validation
- Dry heat tunnel qualification
- Recovery studies
- Method suitability testing
- Equipment performance verification
Challenge materials provide a consistent endotoxin load that enables laboratories to evaluate actual process effectiveness rather than simply assay performance.
Depyrogenation Validation and the 3-Log Reduction Requirement
One of the most common applications for endotoxin challenge materials is depyrogenation validation.
Regulatory guidance typically expects manufacturers to demonstrate at least a 3-log reduction in endotoxin activity.
Example:
| Validation Stage | Endotoxin Level |
|---|---|
| Initial Challenge | 10,000 EU/vial |
| After Dry Heat Treatment | 10 EU/vial |
| Log Reduction | 3-log |
This level of reduction demonstrates effective endotoxin destruction and supports qualification of depyrogenation processes.
For this reason, Endotoxin Challenge Vials remain a standard component of many validation programs.

How FireGene Supports USP <85> and USP <86> Validation
As pharmaceutical quality systems evolve, laboratories increasingly require suppliers that provide more than just reagents.
FireGene supports endotoxin testing and validation workflows through:
Endotoxin Challenge Vials (ECV)
Designed for:
- Depyrogenation validation
- Dry heat oven qualification
- Tunnel validation studies
- Recovery experiments
TAL Endotoxin Test Kits
Supporting traditional USP <85> workflows through:
- Gel-Clot Methods
- Kinetic Chromogenic Methods
- Routine QC Testing
Validation Support Documentation
Including:
- Certificates of Analysis (COAs)
- Technical documentation
- Product specifications
- Validation guidance materials
Global Distribution Support
Providing endotoxin testing solutions to pharmaceutical manufacturers, CROs, CDMOs, and research institutions worldwide.

The Future of Endotoxin Testing
The future of endotoxin testing is not a competition between USP <85> and USP <86>.
Instead, the industry is moving toward a hybrid ecosystem where:
- Traditional TAL/LAL assays provide continuity
- Recombinant technologies expand analytical options
- Validation science becomes increasingly important
- Sustainability considerations influence method selection
Organizations that understand both approaches will be better positioned to adapt to future regulatory and technological developments.
USP <85> vs USP <86>: Validation Checklist
Before implementing recombinant endotoxin testing, validation teams should confirm:
✅ Method suitability completed
✅ Spike recovery demonstrated
✅ Precision established
✅ Accuracy verified
✅ Robustness evaluated
✅ Comparability studies completed
✅ Risk assessment documented
✅ Supplier qualification approved
✅ Regulatory justification prepared
Frequently Asked Questions
Does USP <86> replace USP <85>?
No. USP <85> remains the established compendial standard, while USP <86> provides an alternative recombinant approach.
Is Recombinant Factor C accepted by regulators?
Yes. USP <86> formally recognizes recombinant endotoxin testing methodologies. However, laboratories must still validate suitability for intended applications.
What products are most likely to adopt USP <86>?
Cell therapies, gene therapies, biologics, vaccines, and other advanced therapeutic products are among the earliest adopters.
What is a 3-log endotoxin reduction?
A 3-log reduction represents a 1,000-fold decrease in endotoxin activity. For example, reducing endotoxin levels from 10,000 EU/vial to 10 EU/vial demonstrates successful 3-log reduction.
Why are Endotoxin Challenge Vials used during validation?
They provide a standardized endotoxin load that allows manufacturers to evaluate depyrogenation effectiveness, recovery performance, and process robustness.
Conclusion
USP <85> remains the cornerstone of bacterial endotoxin testing, but USP <86> signals an important evolution toward recombinant, animal-free analytical technologies. Rather than replacing traditional methods, recombinant assays expand the options available to pharmaceutical manufacturers seeking improved sustainability, supply chain resilience, and analytical flexibility.
As biologics, cell therapies, gene therapies, and advanced pharmaceutical products continue to grow, validation teams must understand both compendial frameworks and develop strategies that balance regulatory compliance with scientific innovation.
Organizations that invest today in robust validation programs, qualified suppliers, and comprehensive endotoxin control strategies will be best positioned for the future of pharmaceutical quality control.
FireGene Endotoxin Testing
Ready to run your endotoxin assay?
FireGene offers a complete endotoxin testing toolkit — from TAL reagents and CSE standards to pyrogen-free consumables and LAL reagent water. All products are aligned with USP <85>, EP 2.6.14, and JP 4.01.





