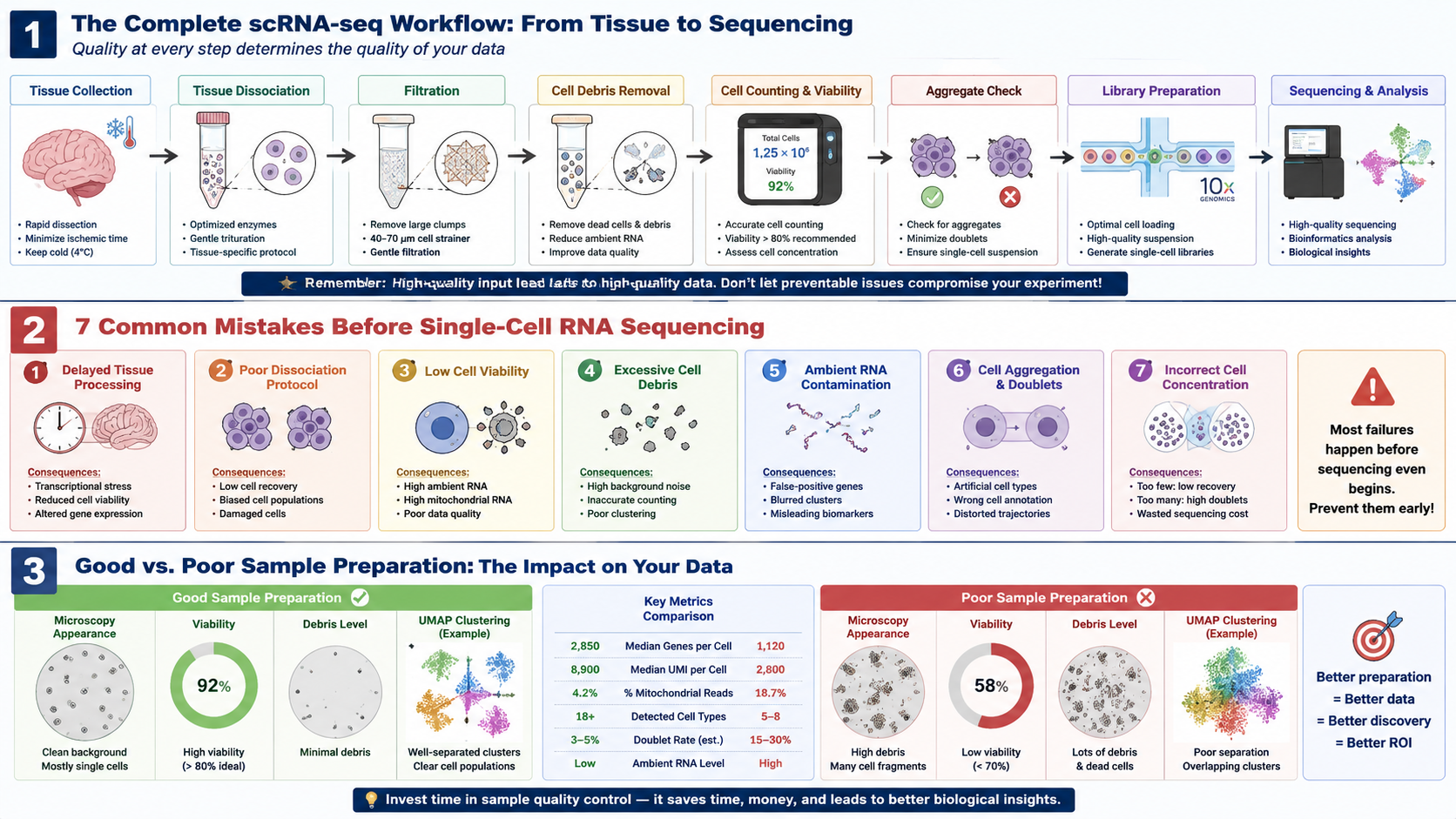
Most Single-Cell RNA Sequencing Problems Begin Long Before Library Preparation
Single-cell RNA sequencing (scRNA-seq) has fundamentally transformed modern biological research. From cancer immunology and neuroscience to developmental biology and precision medicine, researchers can now characterize complex cellular ecosystems with unprecedented resolution.
The rapid adoption of single-cell technologies has fueled ambitious international initiatives such as the Human Cell Atlas, BRAIN Initiative Cell Census Network (BICCN), and numerous large-scale spatial biology programs. More recently, advances in AI-assisted bioinformatics and multiomics have further expanded the impact of single-cell sequencing across both basic research and translational medicine.
Yet despite remarkable improvements in sequencing chemistry, microfluidics, and computational analysis, one reality remains unchanged:
Many single-cell RNA sequencing experiments fail before the sequencing instrument is ever turned on.

Researchers often assume poor sequencing results originate from library preparation, sequencing depth, or bioinformatics analysis. In practice, however, the majority of data quality issues originate much earlier—during tissue collection, cell isolation, sample preparation, and quality control.
Low cell viability.
Excessive cell debris.
Ambient RNA contamination.
Cell aggregation.
Doublets.
Overdigestion.
Improper tissue handling.
Each of these problems can dramatically reduce sequencing quality while remaining invisible until the final dataset has already been generated.
The unfortunate consequence is familiar to many laboratories:
Thousands of dollars invested in sequencing produce data that cannot confidently answer the original biological question.
For this reason, experienced single-cell researchers increasingly recognize that sample preparation—not sequencing—is often the most important determinant of successful scRNA-seq experiments.
The Hidden Cost of Poor Sample Preparation
A single sequencing run may cost thousands of dollars, but the financial investment represents only a small portion of the true expense.
Poor-quality samples also result in:
- Weeks or months of lost research time
- Limited recovery of rare cell populations
- Reduced statistical power
- Failed biological validation
- Delayed publications
- Increased grant costs
- Irreproducible experimental results
Perhaps most importantly, poor sample preparation introduces biological artifacts that cannot be corrected by downstream computational analysis.
No amount of bioinformatics can recover cells that were destroyed during tissue dissociation.
No sequencing platform can distinguish whether inflammatory gene expression reflects true biology or stress induced during sample preparation.
And no computational pipeline can reconstruct rare cell populations that were selectively lost before library preparation.
This is why the phrase "garbage in, garbage out" is especially applicable to single-cell sequencing.
High-quality sequencing always begins with high-quality cells.
What Defines a Successful Single-Cell RNA Sequencing Experiment?
Researchers often judge sequencing success by the number of reads generated or the total number of detected genes.
While these metrics are important, they represent only the final outcome of a much longer experimental workflow.
A successful scRNA-seq experiment actually begins with a high-quality single-cell suspension.
Before sequencing even starts, researchers should already be asking:
- Are the cells healthy?
- Are they truly single cells?
- Has tissue digestion altered native gene expression?
- Is debris minimal?
- Is ambient RNA under control?
- Have fragile cell populations survived processing?
Only when these questions can be answered confidently does sequencing become worthwhile.
High-quality samples generally exhibit several characteristics:
- High cell viability
- Minimal cell aggregation
- Low debris levels
- Low ambient RNA contamination
- Representative recovery of all major cell populations
- Consistent cell concentration
- Limited processing-induced stress responses
These parameters collectively determine whether downstream sequencing accurately reflects the biology of the original tissue.
Mistake #1: Poor Tissue Collection Starts the Failure Cascade
Many researchers focus on optimizing dissociation enzymes or sequencing chemistry while overlooking the very first step of the workflow: tissue collection.
Unfortunately, damage introduced during sample collection cannot be reversed later.
Immediately after tissue excision, cells begin responding to:
- Ischemia
- Temperature fluctuations
- Hypoxia
- Mechanical injury
- Oxidative stress
Within minutes, stress-response genes may become activated, altering transcriptional profiles before tissue processing has even begun.
For fragile tissues such as brain, liver, kidney, and tumors, prolonged delays between collection and processing may substantially reduce cell viability.
Best practices include:
- Minimize ischemic time
- Keep samples appropriately cooled when required
- Standardize collection protocols
- Reduce transportation time
- Process tissues immediately whenever possible
Many laboratories now treat tissue collection as the first quality control checkpoint rather than simply a logistical step.
Mistake #2: Incomplete Tissue Dissociation Leaves Valuable Cells Behind
Releasing individual cells from intact tissue is considerably more complex than simply digesting extracellular matrix proteins.
Each tissue presents unique structural challenges.
Brain tissue contains delicate neuronal networks.
Kidney tissue contains dense tubular structures.
Fibrotic liver samples exhibit excessive extracellular matrix deposition.
Plant tissues possess rigid cell walls.
Tumors often display remarkable heterogeneity across different regions.
Consequently, no universal dissociation protocol exists.
Under-digestion may result in:
- Cell aggregates
- Reduced cell yield
- Increased doublets
- Biased recovery of specific cell populations
Conversely, excessive enzymatic digestion may:
- Damage fragile cell types
- Reduce viability
- Alter transcriptional profiles
- Increase ambient RNA contamination
Optimizing tissue-specific dissociation conditions is therefore essential for preserving native cellular diversity.
Mistake #3: Low Cell Viability Can Distort the Entire Dataset
One of the most widely accepted indicators of sample quality is cell viability. While researchers often focus on sequencing depth or the number of detected genes, poor viability can compromise an experiment before a single droplet is generated.
Dead and dying cells do far more than simply reduce the number of usable cells. They actively release intracellular RNA, DNA, proteins, and cellular debris into the suspension. These contaminants increase background noise and contribute to several downstream issues that negatively affect sequencing quality.
Low viability is commonly associated with:
- Reduced cell recovery
- Increased ambient RNA contamination
- Higher mitochondrial RNA content
- Loss of fragile cell populations
- Poor clustering performance
- Reduced reproducibility across biological replicates
Importantly, poor viability rarely affects every cell type equally.
Neurons, immune cells, endothelial cells, epithelial cells, and stem cells often exhibit markedly different tolerances to enzymatic digestion and mechanical stress. As a result, low-quality sample preparation may selectively eliminate biologically important populations while leaving more resilient cells behind, creating an artificial representation of the original tissue.
This sampling bias cannot be corrected through bioinformatics analysis because the missing cells were never captured.
Researchers should therefore evaluate cell viability immediately after tissue dissociation and again before loading samples onto a droplet-based sequencing platform.
Mistake #4: Cell Debris Is Quietly Destroying Your Sequencing Quality
While most researchers pay close attention to cell viability, cell debris is often underestimated as a major source of sequencing failure.
Every tissue dissociation protocol inevitably generates debris. During enzymatic digestion and mechanical disruption, damaged cells release fragmented membranes, organelles, DNA, proteins, and extracellular RNA into the suspension.
Although these particles may appear insignificant under the microscope, they can substantially affect downstream library preparation.
Excessive debris may contribute to:
- Increased background fluorescence during flow cytometry
- Difficulties in accurate cell counting
- Reduced loading efficiency
- Higher doublet rates
- Increased ambient RNA
- Lower effective sequencing depth
- Reduced clustering accuracy
In highly cellular tissues such as brain, spleen, tumors, and inflamed organs, debris accumulation can become one of the primary factors limiting sequencing quality.
Why Brain Tissue Is Particularly Challenging
Among all mammalian tissues, brain tissue presents one of the greatest challenges for single-cell preparation.
Neurons possess long and delicate cellular processes that are easily disrupted during dissociation. Myelin fragments, lipid-rich debris, and damaged neuronal membranes rapidly accumulate in the suspension, particularly when processing adult or diseased brain tissue.
Researchers working with:
- Alzheimer's disease
- Parkinson's disease
- Glioblastoma
- Stroke
- Multiple sclerosis
- Traumatic brain injury
often encounter exceptionally high levels of cellular debris due to extensive tissue damage and degeneration.
Without effective debris removal, these contaminants can substantially reduce data quality and complicate downstream analyses.
Why Cell Debris Removal Has Become a Standard QC Step
Increasingly, leading single-cell research laboratories now treat cell debris removal as a routine quality control step, rather than an optional refinement.
Removing debris before library preparation offers several advantages:
- Cleaner single-cell suspensions
- Reduced ambient RNA contamination
- Improved cell counting accuracy
- Better droplet encapsulation efficiency
- Higher recovery of intact cells
- Improved clustering and downstream analysis
These benefits are particularly important for challenging tissues, including brain, tumor, fibrotic, and cryopreserved samples.
For laboratories processing delicate or debris-rich tissues, dedicated cleanup workflows can significantly improve overall sequencing performance.
FireGene developed the Brain Tissue Cell Debris Removal Kit to help researchers efficiently remove cellular debris from dissociated brain tissue while preserving intact cells for downstream applications such as:
- Single-cell RNA sequencing
- Single-nucleus RNA sequencing
- Flow cytometry
- Cell sorting
- Spatial transcriptomics
By reducing contaminating debris prior to library preparation, researchers can improve sample purity and increase confidence in downstream biological interpretation.
Mistake #5: Ambient RNA Is the Invisible Enemy of scRNA-seq
One of the most discussed topics in single-cell sequencing over the past several years has been ambient RNA contamination.
Unlike cell debris, ambient RNA cannot be seen under the microscope. Nevertheless, it can profoundly distort sequencing results.
Ambient RNA originates primarily from damaged or lysed cells. During tissue processing, intracellular RNA is released into the surrounding suspension. When droplets are generated during library preparation, this free RNA may be captured alongside intact cells.
The result is an artificial gene expression profile that no longer accurately reflects the biology of the individual cell.
Researchers may observe:
- False-positive gene expression
- Blurred separation between cell clusters
- Misidentification of rare cell populations
- Reduced confidence in marker genes
- Increased computational correction requirements
Recent advances in computational methods—including SoupX, CellBender, and DecontX—have improved the ability to estimate and remove ambient RNA computationally.
However, computational correction should not replace good sample preparation.
Reducing ambient RNA before sequencing remains considerably more effective than attempting to correct contamination afterward.
Maintaining high cell viability, minimizing processing time, optimizing tissue dissociation, and removing cellular debris all contribute to lowering ambient RNA levels before library preparation begins.
Mistake #6: Cell Aggregation and Doublets Reduce Data Accuracy
A common misconception in single-cell sequencing is that every droplet contains exactly one cell. In reality, this assumption depends entirely on the quality of the input sample.
When cells remain attached after tissue dissociation, they may enter the same droplet together, producing what is known as a doublet or multiplet. Instead of representing a single transcriptome, the sequencing data become a composite of two or more cells.
This seemingly small issue can have major consequences.
Doublets may:
- Create artificial cell populations
- Distort cell clustering
- Confuse cell type annotation
- Generate misleading marker genes
- Produce false cell-cell interaction signals
- Bias trajectory and lineage analyses
Although computational tools such as DoubletFinder, Scrublet, and scDblFinder can identify many doublets after sequencing, they cannot recover the true transcriptomes of the original cells. Prevention during sample preparation remains the most effective strategy.
Why Do Cell Aggregates Form?
Several factors contribute to aggregation during tissue processing:
- Incomplete enzymatic digestion
- Excessive extracellular matrix
- Released extracellular DNA from dead cells
- Prolonged sample handling
- Inadequate filtration
- High cell concentrations during processing
Fibrotic tissues and brain samples are particularly prone to aggregation because damaged tissue releases large amounts of extracellular material that promotes cell-cell adhesion.
Simple preventive measures—such as optimizing digestion conditions, gently resuspending cells, filtering through an appropriate mesh size, and removing debris—can substantially reduce aggregation before library preparation.
Mistake #7: Incorrect Cell Concentration Before Loading the Sequencer
Even an excellent single-cell suspension can produce poor sequencing results if the final cell concentration is not properly adjusted.
Droplet-based platforms rely on precise cell loading to maximize the proportion of droplets containing a single cell while minimizing empty droplets and doublets.
Loading too few cells may result in:
- Reduced cell recovery
- Lower sequencing efficiency
- Higher cost per analyzed cell
Loading too many cells increases the probability that multiple cells enter the same droplet, leading to:
- Elevated doublet rates
- More complex downstream filtering
- Reduced confidence in biological interpretation
Accurate cell counting is therefore a critical quality control step.
Researchers should evaluate:
- Total cell concentration
- Cell viability
- Aggregate frequency
- Debris level
- Suspension uniformity
Only after confirming these parameters should samples proceed to library preparation.
Build a Sample Preparation QC Workflow Before Sequencing
Successful scRNA-seq experiments do not rely on a single optimization step. Instead, they result from a standardized workflow in which each stage is monitored using objective quality control criteria.
A practical quality control workflow may include:
Fresh Tissue Collection
│
▼
Rapid Transport Under Optimized Conditions
│
▼
Tissue-Specific Dissociation
│
▼
Cell Filtration
│
▼
Cell Debris Removal
│
▼
Cell Counting
│
▼
Cell Viability Assessment
│
▼
Aggregate Inspection
│
▼
Optimize Cell Concentration
│
▼
Single-Cell Library Preparation
│
▼
SequencingRather than treating quality control as a final checkpoint, leading laboratories integrate QC throughout the entire workflow. Identifying problems before sequencing is far more efficient than attempting to rescue compromised datasets afterward.
For researchers processing challenging brain tissues, incorporating a dedicated debris removal step before cell counting and library preparation can significantly improve sample quality.
A Practical Checklist Before Sending Samples for Sequencing
Before submitting samples for sequencing, consider the following questions:
✅ Was the tissue processed promptly after collection?
✅ Was the dissociation protocol optimized for the specific tissue type?
✅ Are cell viability and recovery acceptable?
✅ Has excessive cellular debris been removed?
✅ Are aggregates or clumps absent?
✅ Has the sample been filtered appropriately?
✅ Is the cell concentration within the recommended range for your sequencing platform?
✅ Has ambient RNA been minimized as much as possible?
If any of these questions cannot be answered confidently, additional optimization before sequencing may save substantial time, cost, and effort.
Case Study: Why Two Laboratories Obtained Completely Different scRNA-seq Results from the Same Tissue
Imagine two research laboratories studying the same biological question.
Both groups receive freshly collected mouse brain tissue.
Both use the same sequencing platform.
Both target approximately 10,000 cells.
Both follow nearly identical library preparation protocols.
Yet after sequencing, their datasets look completely different.
Laboratory A identifies more than twenty distinct cellular populations, including rare microglial subsets and neural progenitor cells.
Laboratory B detects only a limited number of broad cell clusters with poor separation and substantially fewer recovered cells.
What happened?
The sequencing platform was not responsible.
Neither was the bioinformatics pipeline.
The difference originated during sample preparation.
Laboratory A
The first laboratory implemented a standardized sample preparation workflow that emphasized quality control before sequencing.
Their protocol included:
- Rapid tissue processing immediately after collection
- Optimized tissue-specific enzymatic dissociation
- Gentle mechanical handling
- Removal of cellular debris
- Cell filtration
- Viability assessment
- Cell counting before library preparation
As a result, the final suspension contained:
- High viability
- Minimal debris
- Low aggregate frequency
- Reduced ambient RNA
- Uniform single-cell suspension
The sequencing data demonstrated:
- Excellent cluster separation
- High median genes per cell
- Low mitochondrial RNA
- Robust recovery of rare neuronal populations
Laboratory B
The second laboratory focused primarily on sequencing throughput.
Although tissue dissociation was completed successfully, several quality control steps were omitted.
Specifically:
- Tissue remained at room temperature longer before processing.
- Cellular debris was not removed.
- Cell viability was not evaluated before loading.
- Cell aggregates were only partially filtered.
- Cell concentration was estimated rather than accurately measured.
Following sequencing, several quality issues became apparent.
Researchers observed:
- Increased background RNA
- Poor clustering
- Higher doublet frequency
- Lower cell recovery
- Elevated mitochondrial gene expression
- Reduced detection of rare cell populations
Although sequencing depth was comparable to Laboratory A, the biological conclusions were substantially weaker.
The Lesson
This scenario reflects one of the most common misconceptions in single-cell sequencing:
Researchers often believe sequencing quality is determined by the sequencing platform.
In reality,
sample quality determines sequencing quality.
Library preparation and sequencing can only capture the biological information present in the input sample.
If viable cells have already been lost—or if debris, ambient RNA, and aggregates dominate the suspension—no sequencing chemistry or computational algorithm can fully restore the missing biological signal.
Prevention Is Far More Effective Than Correction
Today, computational tools such as:
- SoupX
- CellBender
- DecontX
- DoubletFinder
have significantly improved the ability to identify technical artifacts after sequencing.
However, these algorithms were developed to reduce technical noise—not to replace good laboratory practice.
The most reliable strategy remains preventing these issues before sequencing begins.
By standardizing tissue handling, optimizing dissociation conditions, removing cellular debris, and implementing routine quality control, researchers can dramatically improve both sequencing performance and experimental reproducibility.
For laboratories processing challenging tissues—particularly adult brain, tumor, inflamed tissue, or archived specimens—incorporating a dedicated debris removal step before library preparation can further enhance sample quality and reduce downstream technical artifacts.
Researchers seeking reliable Research Reagents and Sample Preparation Solutions increasingly recognize that high-quality sequencing begins not with the sequencer, but with careful preparation of the biological sample.
7 Critical Checkpoints Before Every Single-Cell RNA Sequencing Experiment
Before sending your samples for library preparation, take a few minutes to review the following checklist. Addressing these seven checkpoints can significantly improve data quality and reduce the risk of failed sequencing runs.
| Checkpoint | Why It Matters | Potential Consequence if Ignored |
|---|---|---|
| 1. Rapid Tissue Processing | Preserves native gene expression and cell viability. | Stress-induced transcriptional changes and reduced cell recovery. |
| 2. Optimized Tissue Dissociation | Generates a representative single-cell suspension without excessive damage. | Cell loss, poor recovery, and biased cell populations. |
| 3. High Cell Viability | Minimizes ambient RNA contamination and improves sequencing accuracy. | Increased background RNA, lower-quality libraries, and distorted biological signals. |
| 4. Cell Debris Removal | Removes damaged cellular fragments that interfere with downstream workflows. | Higher background noise, inaccurate cell counting, and reduced clustering performance. |
| 5. Aggregate and Doublet Control | Ensures each droplet contains a single cell whenever possible. | Artificial hybrid cell populations and inaccurate cell annotation. |
| 6. Accurate Cell Counting and Concentration | Optimizes loading efficiency for droplet-based platforms. | Low recovery or excessive doublets caused by incorrect loading concentrations. |
| 7. Final Quality Control Review | Confirms the sample is ready for sequencing. | Costly sequencing failures that could have been prevented. |
A Successful scRNA-seq Experiment Starts Here
The journey from tissue collection to high-quality sequencing data is not defined by a single step. Instead, success depends on a series of carefully controlled decisions made before the sample ever reaches the sequencing instrument.
Researchers who consistently produce high-quality datasets share one common principle:
They treat sample preparation as an integral part of sequencing—not merely a preliminary step.
By implementing standardized quality control procedures, monitoring cell viability, reducing debris, minimizing ambient RNA contamination, and optimizing tissue dissociation, laboratories can dramatically improve sequencing outcomes and generate datasets that more accurately reflect true biology.
For researchers working with brain tissue and other debris-rich samples, incorporating a dedicated cleanup step before library preparation can further improve sample purity and support more reliable downstream analyses.
Learn more about the FireGene Brain Tissue Cell Debris Removal Kit for single-cell and single-nucleus sequencing workflows:
📋 Key Takeaways
- Most scRNA-seq failures occur before sequencing begins, during tissue collection and sample preparation.
- Cell viability, debris removal, and aggregate control are among the most critical determinants of sequencing success.
- Ambient RNA contamination cannot be fully corrected computationally—prevention is far more effective.
- Standardized quality control workflows improve reproducibility and maximize the return on sequencing investments.
- High-quality sample preparation is the foundation of high-quality biological discovery.
The Real Cost of a Failed Single-Cell RNA Sequencing Experiment
When researchers think about the cost of a failed single-cell RNA sequencing experiment, they often focus on sequencing expenses alone.
In reality, sequencing represents only one component of a much larger investment.
By the time a sample reaches the sequencing platform, weeks—or even months—of work have already been completed.
Researchers have invested significant time in:
- Experimental design
- Animal breeding or clinical sample collection
- Tissue harvesting
- Cell isolation
- Reagent preparation
- Sample quality assessment
- Library construction
If the sample quality is poor, the entire downstream workflow is compromised regardless of sequencing depth or bioinformatics sophistication.
The Financial Impact Goes Beyond Sequencing
A failed scRNA-seq experiment often results in far greater losses than the sequencing invoice itself.
Additional costs may include:
- Experimental animals
- Clinical sample acquisition
- Cell culture reagents
- Tissue dissociation reagents
- Sequencing library preparation kits
- Sequencing services
- Bioinformatics analysis
- Research personnel time
For laboratories processing precious human biopsies, patient-derived xenografts (PDX), or rare disease samples, failed sequencing may not simply require repeating an experiment—it may be impossible to repeat at all.
Some biological specimens are available only once.
In these situations, poor sample preparation can result in the permanent loss of irreplaceable biological information.
Lost Time Is Often the Greatest Expense
While financial costs are significant, time is frequently the most valuable resource.
A failed sequencing experiment may delay:
- Manuscript submission
- Grant milestones
- Drug discovery programs
- Student graduation timelines
- Collaborative research projects
Repeating tissue collection, sample preparation, library construction, and sequencing can extend project timelines by several weeks or even months.
For competitive research fields such as neuroscience, cancer biology, immunology, and spatial transcriptomics, these delays may have substantial scientific and strategic consequences.
Sample Quality Determines Return on Investment
Modern sequencing platforms have become increasingly powerful.
Today's instruments routinely generate millions of sequencing reads with exceptional accuracy.
However, sequencing technology cannot compensate for poor biological input.
If the starting sample contains:
- Low cell viability
- Excessive cellular debris
- High ambient RNA contamination
- Cell aggregates
- Doublets
- Processing-induced transcriptional stress
then even the highest sequencing depth cannot recover the original biological information.
In other words,
the return on investment of every sequencing experiment is ultimately determined by sample quality.
Small Improvements Before Sequencing Can Prevent Major Losses
Fortunately, many of the most common causes of sequencing failure are preventable.
Simple quality control measures performed before library preparation can substantially improve downstream performance.
These include:
- Standardizing tissue collection procedures
- Optimizing tissue-specific dissociation protocols
- Minimizing processing time
- Monitoring cell viability
- Removing cellular debris
- Filtering aggregates
- Verifying cell concentration before loading
Although each individual step requires only a modest amount of additional time, together they can dramatically improve sequencing success and reduce the likelihood of costly experimental repetition.
For debris-rich tissues such as adult brain, inflamed tissue, and certain tumor specimens, incorporating a dedicated cleanup step before sequencing may further improve sample quality and downstream reproducibility.
FireGene's Brain Tissue Cell Debris Removal Kit was developed to support this critical stage of the workflow by helping researchers generate cleaner cell suspensions for single-cell and single-nucleus sequencing applications.
One Final Thought
As sequencing technologies continue to become faster, more sensitive, and more affordable, the primary limitation is no longer the sequencer.
Instead, it is the quality of the biological sample entering the workflow.
A laboratory may invest thousands of dollars in sequencing and computational analysis, but a relatively small investment in sample quality control can determine whether that investment produces a publication-ready dataset—or a failed experiment.
In many cases, the most cost-effective improvement a laboratory can make is not upgrading its sequencer, but optimizing its sample preparation workflow.
The Future of Single-Cell Sample Preparation
As single-cell technologies continue to evolve, the importance of sample preparation is becoming even more apparent.
Emerging applications—including single-cell multiomics, spatial transcriptomics, and AI-assisted cell atlas construction—place even greater demands on sample quality.
Researchers increasingly require suspensions that preserve not only RNA integrity but also native cellular states and rare cell populations. This shift has transformed sample preparation from a routine laboratory procedure into a critical determinant of experimental success.
Several trends are shaping the future of the field:
Single-Nucleus Sequencing
Single-nucleus RNA sequencing (snRNA-seq) has become the preferred approach for many frozen, fibrotic, and difficult-to-dissociate tissues, expanding opportunities for archived clinical samples and complex organs.
Spatial Transcriptomics Integration
As spatial technologies become more widespread, researchers increasingly combine spatial transcriptomics with scRNA-seq or snRNA-seq datasets. High-quality cell or nuclei isolation remains essential for generating accurate reference atlases and validating spatial findings.
Multiomics Workflows
Modern platforms can simultaneously profile RNA, chromatin accessibility, proteins, and immune receptor repertoires. These applications require exceptionally clean, viable samples because poor preparation affects multiple molecular layers simultaneously.
AI-Assisted Quality Control
Machine learning is beginning to support automated assessment of cell viability, debris content, and sample quality. While these tools are promising, they complement rather than replace robust laboratory workflows.
Ultimately, regardless of advances in sequencing chemistry or computational analysis, one principle remains unchanged:
The quality of single-cell sequencing data is determined first by the quality of the sample.
Researchers who invest in standardized tissue processing, optimized dissociation, effective debris removal, and rigorous quality control are far more likely to generate reproducible datasets and meaningful biological insights.
References
- Stuart T, Satija R. Integrative single-cell analysis. Nature Reviews Genetics. 2019;20:257–272.
- Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis. Molecular Systems Biology. 2019;15:e8746.
- Mereu E, et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nature Biotechnology. 2020;38:747–755.
- Vieth B, Parekh S, Ziegenhain C, et al. A systematic evaluation of single-cell RNA-sequencing analysis pipelines. Nature Communications. 2019;10:4667.
- Young MD, Behjati S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience. 2020;9(12):giaa151.
- Fleming SJ, Marioni JC, Babadi M. CellBender removes background noise from single-cell RNA sequencing data. Nature Methods. 2023.
- McGinnis CS, Murrow LM, Gartner ZJ. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Systems. 2019;8(4):329–337.
- Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biology. 2019;20:296.
- Regev A, et al. The Human Cell Atlas. eLife. 2017;6:e27041.
FireGene Single-Cell Sample Prep
Looking for validated dissociation kits?
FireGene's tissue dissociation kits are optimized for specific organ types — brain, tumor, liver, GI, reproductive, and more. Validated for 10x Genomics Chromium and BD Rhapsody workflows.





