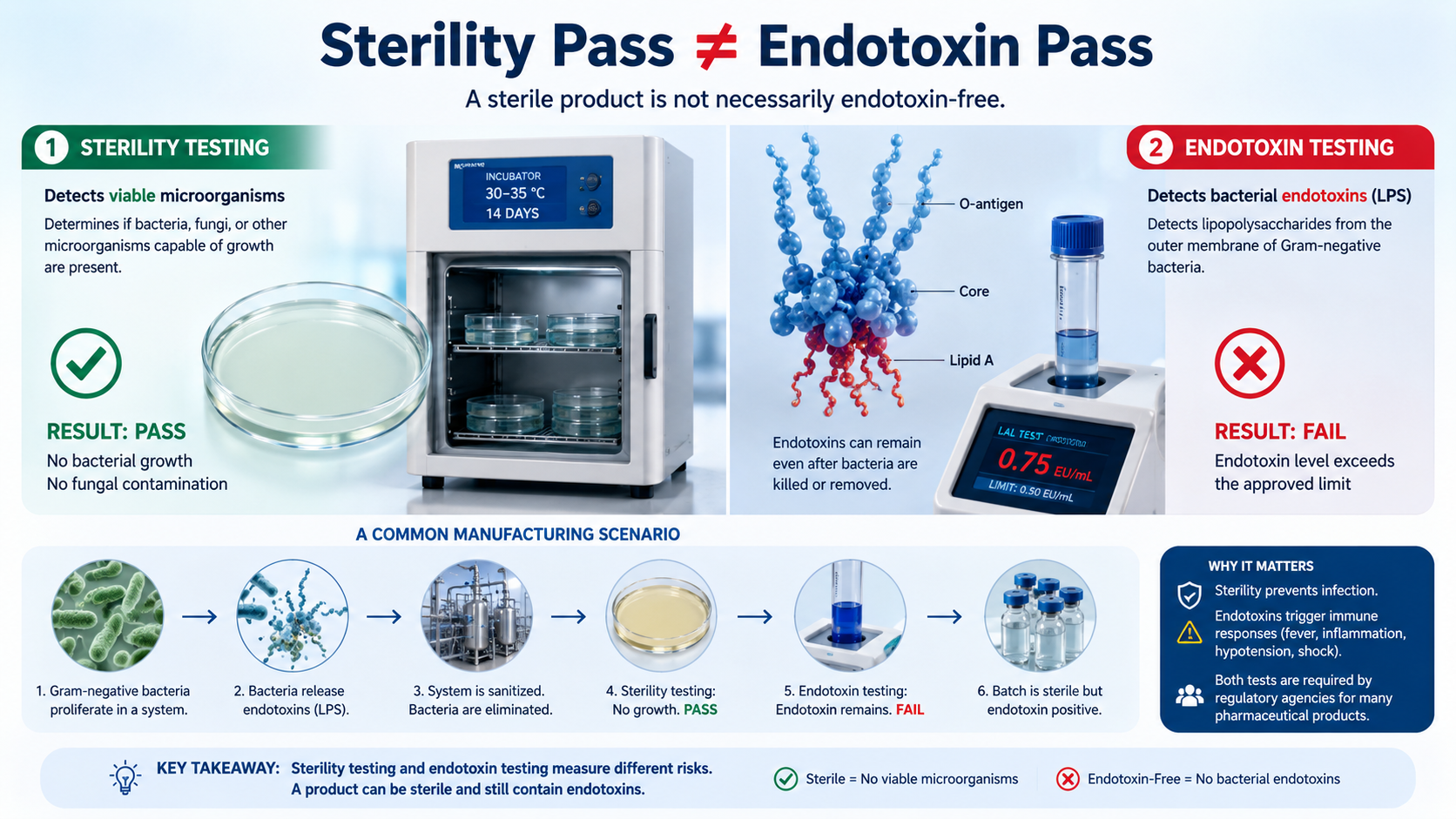
A batch passes sterility testing.
No bacterial growth is detected. No fungal contamination is observed. All microbiological indicators suggest the product is suitable for release.
Then the endotoxin results arrive.
The batch fails.
For many professionals working in pharmaceutical manufacturing, biologics production, medical device development, or cell and gene therapy, this situation can seem counterintuitive. If a product is sterile, how can it still fail bacterial endotoxin testing?
The answer lies in a critical distinction that is often misunderstood throughout the life sciences industry: sterility and endotoxin control address two fundamentally different risks.
A sterile product is not necessarily endotoxin-free.
In fact, endotoxins can remain present and biologically active even after the microorganisms that produced them have been destroyed. This is why global regulatory agencies require both sterility testing and bacterial endotoxin testing for many pharmaceutical products.
As modern therapies become increasingly complex and patient populations become more vulnerable, understanding the difference between microbial contamination and endotoxin contamination has become more important than ever.
Sterility and Endotoxin Testing Measure Different Risks
Sterility testing and endotoxin testing are often discussed together because both are critical for product safety. However, they evaluate entirely different quality attributes.
Sterility testing is designed to determine whether viable microorganisms are present in a product. A passing sterility result demonstrates that bacteria, fungi, or other microorganisms capable of growth are absent under the test conditions.
Endotoxin testing serves a different purpose. Rather than looking for living organisms, it detects bacterial endotoxins, specifically lipopolysaccharides (LPS) derived from the outer membrane of Gram-negative bacteria.
This distinction is important because endotoxins can remain in a product long after the bacteria themselves have been eliminated.
A product may therefore:
- Pass sterility testing
- Contain no viable microorganisms
- Still fail bacterial endotoxin testing
This is not a contradiction. It is simply the result of measuring two different contamination risks.
From a patient safety perspective, both risks matter. Microbial contamination can cause infection, while endotoxin contamination can trigger fever, inflammation, hypotension, and in severe cases, shock-like reactions.
What Makes Endotoxins Different?
Endotoxins are naturally occurring components of the outer membrane of Gram-negative bacteria such as Escherichia coli, Pseudomonas aeruginosa, Enterobacter species, and Klebsiella species.
When these microorganisms grow, die, or undergo cell lysis, endotoxins are released into the surrounding environment.
Unlike living bacteria, endotoxins are not capable of reproduction. They do not grow, multiply, or spread on their own.
However, they possess another characteristic that makes them particularly challenging from a quality-control perspective: they are remarkably stable.
Many contaminants can be eliminated through conventional sterilization processes. Endotoxins often cannot.
This is the key reason why a sterile product can still pose a pyrogen risk.
Why Sterilization Does Not Necessarily Remove Endotoxins
One of the most common misconceptions in pharmaceutical manufacturing is the assumption that sterilization destroys everything that could potentially harm a patient.
In reality, sterilization processes are specifically designed to eliminate viable microorganisms. They are not necessarily intended to remove endotoxins.
Steam sterilization, sterile filtration, gamma irradiation, and other common sterilization methods are highly effective at reducing microbial contamination. However, endotoxins can survive conditions that readily destroy bacterial cells.
Consider a typical autoclave cycle. Temperatures around 121°C are sufficient to kill most microorganisms. Yet endotoxins may remain biologically active after exposure to those same conditions.
This explains why a product can emerge from sterilization free of viable bacteria while still containing measurable endotoxin levels.
For manufacturers, this distinction has significant implications. It means that sterility assurance alone cannot guarantee product safety, particularly for injectable products that come into direct contact with the bloodstream.
A Common Manufacturing Scenario: Sterile but Endotoxin Positive
To understand how this occurs in practice, consider a pharmaceutical water system.
A small population of Gram-negative bacteria develops within part of the distribution loop. Over time, the bacteria proliferate and release endotoxins into the system.
Eventually, the contamination is detected. The system is sanitized, and the microbial population is eliminated.
Subsequent microbiological testing confirms that viable bacteria are no longer present.
At this point, many people would assume the problem has been resolved.
However, the endotoxins generated during the contamination event may still remain within the system.
Water from that system is later used in manufacturing.
The resulting batch passes sterility testing because no viable microorganisms are present.
The same batch fails bacterial endotoxin testing because endotoxins remain.
This type of scenario is one of the most common root causes of unexpected endotoxin failures across the pharmaceutical industry.
Why Endotoxin Contamination Matters
Unlike microbial contamination, endotoxins do not cause infection.
Instead, they trigger powerful immune responses.
When endotoxins enter the bloodstream, immune cells recognize them as danger signals and rapidly initiate inflammatory pathways. Cytokines and other mediators are released, producing a cascade of biological responses.
Depending on the amount of endotoxin present and the patient's condition, symptoms may include:
- Fever
- Chills
- Headache
- Malaise
- Hypotension
- Systemic inflammation
In severe situations, endotoxin exposure can result in shock-like symptoms and significant clinical complications.
This is why endotoxin limits are tightly regulated for injectable pharmaceuticals, biologics, vaccines, dialysis products, and many medical devices.
Even when a product is completely sterile, excessive endotoxin levels can still create unacceptable patient risk.
Depyrogenation Is Not the Same as Sterilization
Another common source of confusion is the relationship between sterilization and depyrogenation.
Although the terms are sometimes used interchangeably in casual conversation, they describe different objectives.
Sterilization is intended to eliminate viable microorganisms.
Depyrogenation is intended to destroy or remove pyrogenic substances such as bacterial endotoxins.
A process capable of achieving sterility may not necessarily achieve depyrogenation.
This distinction becomes particularly important when evaluating manufacturing equipment, glassware, containers, and critical process components.
For example, a depyrogenation tunnel used for pharmaceutical glass containers operates under conditions specifically validated for endotoxin reduction. The objective is not simply to eliminate microorganisms but to reduce endotoxin burden to acceptable levels.
Understanding the difference between these two processes is essential for designing an effective contamination-control strategy.
Why Endotoxin Control Has Become More Challenging in Modern Biopharmaceutical Manufacturing
The pharmaceutical industry is changing rapidly.
Twenty years ago, many products consisted primarily of relatively simple small-molecule formulations. Today, manufacturers routinely develop monoclonal antibodies, recombinant proteins, antibody-drug conjugates, viral vectors, cell therapies, and gene therapies.
These products introduce new challenges for endotoxin control.
Biologically derived raw materials often carry higher contamination risks than traditional synthetic ingredients. Manufacturing processes are more complex, supply chains are longer, and product sensitivity has increased dramatically.
At the same time, regulatory expectations continue to evolve.
Quality systems are increasingly expected to demonstrate proactive contamination prevention rather than relying solely on final product testing.
As a result, endotoxin control is no longer viewed as a single release test performed at the end of manufacturing. It has become an integrated strategy spanning raw material qualification, process development, environmental monitoring, water system management, cleaning validation, and finished-product testing.
Organizations that treat endotoxin testing as a standalone activity often find themselves reacting to failures rather than preventing them.
Special Considerations for Cell and Gene Therapy Products
Few sectors illustrate this challenge more clearly than cell and gene therapy manufacturing.
Cell and gene therapy products frequently involve highly complex biological materials, short production timelines, and patient-specific manufacturing approaches.
Potential endotoxin sources may include:
- Viral vectors
- Plasmid DNA
- Cell culture media
- Cytokines
- Growth factors
- Single-use processing systems
Unlike many traditional pharmaceuticals, these products often cannot undergo terminal sterilization after manufacturing.
Consequently, contamination prevention becomes critically important.
A single endotoxin excursion may compromise an entire manufacturing campaign, delay patient treatment, or trigger extensive investigations.
For this reason, endotoxin control strategies are increasingly being incorporated into every stage of cell and gene therapy process development, from raw material selection through final product release.
Building an Effective Endotoxin Control Strategy
The most successful manufacturers focus on prevention rather than detection alone.
While bacterial endotoxin testing remains essential, testing should serve as one component of a broader contamination-control framework.
An effective endotoxin control strategy typically includes:
Water System Management
Water systems remain one of the most common sources of endotoxin contamination. Routine monitoring, trend analysis, sanitization, and biofilm control are critical.
Supplier Qualification
Understanding the endotoxin risk associated with incoming materials helps reduce contamination before manufacturing begins.
Cleaning Validation
Validated cleaning procedures should demonstrate effective removal of both product residues and endotoxins.
Environmental Monitoring
Although environmental monitoring does not directly measure endotoxins, it can provide early warning signs of Gram-negative bacterial contamination.
In-Process Testing
Monitoring critical process steps allows manufacturers to identify contamination events before they affect finished product quality.
Routine Endotoxin Testing
Validated bacterial endotoxin testing methods based on TAL/LAL Reagent technology remain the industry standard for detecting and quantifying endotoxin contamination.
Together, these activities create multiple layers of protection throughout the manufacturing lifecycle.
The Continuing Role of TAL/LAL Reagent-Based Endotoxin Testing
Despite advances in pharmaceutical manufacturing, bacterial endotoxin testing remains a cornerstone of quality control.
Methods based on TAL/LAL Reagent technology continue to be recognized globally under USP <85>, European Pharmacopoeia 2.6.14, and related regulatory standards.
Depending on laboratory requirements, organizations may utilize:
- Gel Clot Methods for simple qualitative testing
- Kinetic Chromogenic Methods for quantitative analysis and high-throughput applications
- Kinetic Turbidimetric Methods for automated endotoxin measurement
The choice of methodology depends on product characteristics, throughput requirements, sensitivity needs, and regulatory expectations.
Regardless of the method selected, endotoxin testing remains one of the most important safeguards protecting patients from pyrogen-related adverse reactions.
Conclusion
The assumption that a sterile product is automatically endotoxin-free remains one of the most persistent misconceptions in pharmaceutical manufacturing.
Sterility testing and endotoxin testing address different risks, measure different contaminants, and provide different information about product safety.
A product can contain no viable microorganisms and still harbor biologically active endotoxins capable of triggering significant patient reactions.
As pharmaceutical products become more complex and manufacturing processes become more sophisticated, understanding this distinction becomes increasingly important.
Effective contamination control requires more than simply achieving sterility. It requires a comprehensive strategy that addresses microbial contamination, endotoxin contamination, process design, raw material quality, and ongoing monitoring throughout the product lifecycle.
Only by controlling both sterility and endotoxin risk can manufacturers ensure the safety, quality, and regulatory compliance of the products delivered to patients worldwide.
About FireGene Endotoxin Testing Solutions
FireGene provides a comprehensive portfolio of endotoxin testing solutions designed to support pharmaceutical, biotechnology, medical device, and cell and gene therapy applications.
Our portfolio includes Gel Clot TAL/LAL Reagent, Kinetic Chromogenic Endotoxin Test Kits, Control Standard Endotoxin (CSE), pyrogen-free consumables, and technical support for method development and validation.
Whether you are establishing a new endotoxin testing program or optimizing an existing quality-control workflow, FireGene can help support reliable and compliant bacterial endotoxin testing throughout the product lifecycle.
FireGene Endotoxin Testing
Ready to run your endotoxin assay?
FireGene offers a complete endotoxin testing toolkit — from TAL reagents and CSE standards to pyrogen-free consumables and LAL reagent water. All products are aligned with USP <85>, EP 2.6.14, and JP 4.01.





