Next-generation sequencing is one of the most powerful and useful technologies in modern genomics. It helps researchers study DNA and RNA with speed, depth, and accuracy. From basic life science research to gene expression studies, biomarker discovery, oncology research, and precision medicine, NGS provides valuable insights into biological systems.
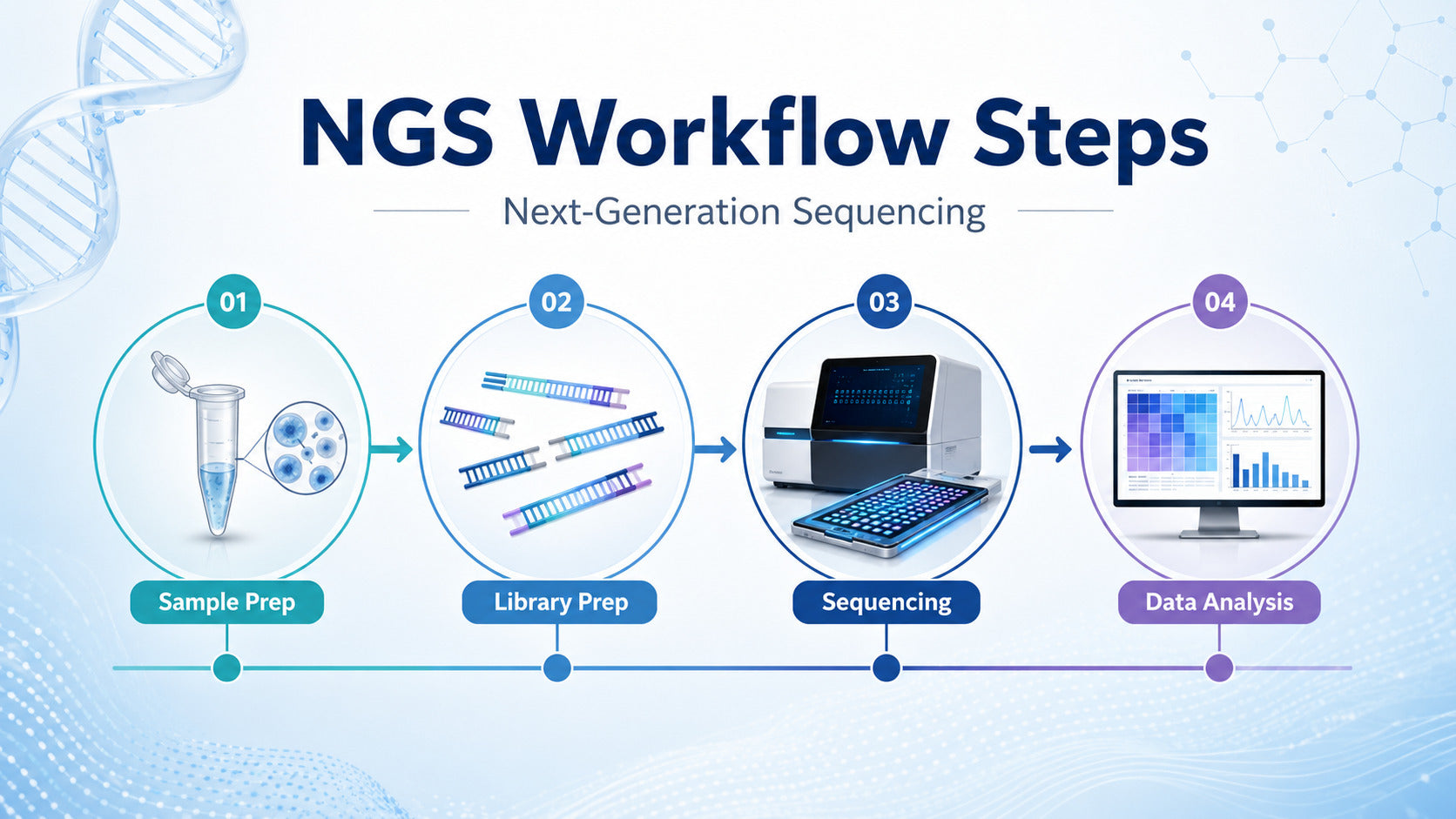
For beginners, the NGS process becomes easier when it is understood step by step. A typical next-generation sequencing workflow includes four main stages: nucleic acid extraction, library preparation, sequencing, and data analysis. Each stage contributes to the quality and reliability of the final result. This guide explains the complete NGS sequencing workflow in a clear, practical, and research-focused way.
What Is an NGS Workflow?
An NGS workflow is the complete process used to move from a biological sample to meaningful sequencing results. It begins with extracting nucleic acids from a sample, preparing those nucleic acids into sequencing-ready libraries, running the libraries on a sequencing platform, and then analyzing the generated data. In simple words, the NGS workflow turns DNA or RNA into readable digital sequence information. This information can then be used to identify genetic variants, study gene expression, compare samples, explore biological pathways, and support advanced research decisions.
A successful workflow depends on careful planning at every stage. High-quality samples, well-prepared libraries, suitable sequencing depth, and reliable NGS data analysis all work together to support accurate and useful results.
Why Understanding NGS Workflow Steps Matters
NGS is a highly precise technology, and each step adds value to the final output. When the sample is prepared carefully and the library is built correctly, researchers can achieve strong sequencing performance and meaningful data.
Understanding the full NGS sequencing workflow helps researchers:
- Choose the right sample preparation method
- Improve nucleic acid quality before sequencing
- Select the right library preparation approach
- Plan read length, depth, and coverage more effectively
- Support clear and confident data interpretation
- Build a smoother workflow from sample to result
This is especially helpful for NGS for beginners because it gives a simple roadmap from starting material to final biological insight.
Step 1: Nucleic Acid Extraction for NGS
The first step in the NGS workflow is nucleic acid extraction. This step involves isolating DNA or RNA from biological samples such as blood, tissue, cells, biofluids, FFPE samples, cfDNA samples, or single cells. The goal of nucleic acid extraction for NGS is to obtain genetic material that is suitable for library preparation. Strong extraction supports good yield, clean samples, and reliable downstream sequencing results.
DNA and RNA Isolation
NGS can begin with different types of nucleic acids depending on the research goal. For DNA sequencing, genomic DNA, cfDNA, or targeted DNA regions may be used. For RNA sequencing, total RNA or mRNA is commonly isolated and converted into cDNA before library preparation. The extraction method should match the sample type. Fresh tissue, blood, FFPE tissue, and low-input samples may require different extraction strategies. Choosing the right isolation method helps preserve sample quality and supports better downstream performance.
Yield, Purity, and Quality
Three things are especially important after extraction: yield, purity, and quality. Yield means the amount of DNA or RNA recovered from the sample. A suitable yield gives the library preparation process enough input material.
Purity means the nucleic acid is clean and ready for enzymatic reactions used during library preparation. Quality means the DNA or RNA has the right integrity for the selected sequencing application. High-quality DNA supports many DNA sequencing workflows, while RNA integrity is especially valuable for RNA-Seq.
QC After Nucleic Acid Extraction
After extraction, a quality control step helps confirm that the sample is ready for the next stage. Common QC methods include UV spectrophotometry, fluorometric quantification, gel electrophoresis, and microfluidic electrophoresis.
UV spectrophotometry can help assess purity using A260/280 and A260/230 ratios. Fluorometric methods are useful for accurate DNA or RNA quantification. Electrophoresis-based methods can show fragment size distribution and nucleic acid integrity. This QC step gives researchers confidence before moving into the NGS library preparation process.
Step 2: NGS Library Preparation Process
The second step is the NGS library preparation process. In this step, purified DNA or cDNA is converted into a sequencing-ready library. An NGS library is a collection of DNA fragments prepared with special adapter sequences. These adapters allow the fragments to bind to the sequencing platform and be read during sequencing.
Library preparation is one of the most important steps in the next-generation sequencing workflow because it directly supports sequencing quality, read distribution, coverage, and data output.
Fragmentation
In many workflows, DNA or cDNA is fragmented into smaller pieces before sequencing. Fragmentation can be mechanical, enzymatic, or transposase-based, depending on the library preparation method.
The ideal fragment size depends on the sequencing platform, read length, and application. Whole genome sequencing, RNA sequencing, targeted sequencing, and cfDNA sequencing may require different fragment size ranges.
Adapter Ligation
After fragmentation, adapters are attached to both ends of the DNA fragments. These adapters contain sequences needed for sequencing, amplification, and sample identification. For Illumina-based workflows, adapters commonly include sequences compatible with flow cell binding and sequencing primers. Proper adapter ligation helps the library interact correctly with the sequencer.
Indexing, Barcoding, and Multiplexing
Indexes or barcodes can be added during library preparation. These short, unique sequences help identify which reads belong to which sample. This allows multiple samples to be pooled and sequenced together in the same run. This process is called multiplexing. It saves time, improves workflow efficiency, and helps researchers make better use of sequencing capacity.
Library Amplification and Cleanup
Some workflows include PCR amplification to increase the amount of the prepared library. This is especially useful when the starting material is limited. Cleanup steps also help refine the library by keeping the desired fragment range. A well-cleaned and well-prepared library supports smooth sequencing and high-quality data generation.
Library Quantification and QC
Before sequencing, libraries should be quantified and checked for size distribution. Library quantification helps ensure that the correct amount of library is loaded onto the sequencer. This step supports consistent data output, balanced sample pooling, and strong sequencing performance.
FireGene provides NGS-related solutions that support researchers during library preparation and workflow planning. With suitable NGS Library Prep Kits and molecular biology reagents, researchers can move from sample preparation to sequencing-ready libraries with confidence.
Optional Step: Target Enrichment
In some NGS workflows, researchers do not need to sequence the whole genome. Instead, they may only want to study selected genes, panels, exons, or genomic regions. This is where target enrichment becomes useful. Target enrichment helps focus sequencing on specific regions of interest. It can improve coverage of important targets, support efficient sequencing design, and make data analysis more focused.
Amplicon-Based Enrichment
Amplicon sequencing uses PCR primers to amplify selected regions before sequencing. This approach is often used for small panels, mutation analysis, microbial studies, and targeted research applications.
Hybridization Capture
Hybridization capture uses probes to pull down selected genomic regions from a prepared library. This approach is commonly used for larger panels, exome sequencing, and more complex targeted sequencing applications.
When Targeted Sequencing Is Useful
Targeted sequencing is useful when researchers need deeper coverage of selected regions instead of broad whole-genome data. It is often chosen for oncology research, inherited disease studies, biomarker discovery, and clinical research workflows.
Step 3: Sequencing
The third step in the NGS workflow is sequencing. In this stage, the prepared library is loaded onto an NGS platform, where the DNA fragments are read base by base. Different sequencing platforms use different technologies, but the goal is the same: to generate sequence reads that represent the DNA or RNA content of the original sample.
Cluster Generation
In many short-read sequencing workflows, library fragments bind to a flow cell and are amplified to form clusters. Each cluster contains many copies of a single library fragment. This amplification makes the signal strong enough for the sequencer to detect. Cluster generation is a key part of the sequencing process because cluster quality supports read quality, data output, and sequencing accuracy.
Sequencing by Synthesis Technology
Sequencing by synthesis technology is widely used in NGS. In this method, nucleotides are added one at a time to a growing DNA strand. Each incorporated base is detected, allowing the system to determine the sequence of the fragment. This technology enables millions or billions of DNA fragments to be sequenced in parallel. That is why next-generation sequencing is much faster and more powerful than traditional sequencing methods.
Read Length, Depth, and Coverage
Read length refers to the number of bases read from each DNA fragment. Shorter or longer read lengths may be selected depending on the application.Sequencing depth refers to how many times a region is read. Higher depth can support confident analysis of variants, complex samples, and targeted regions. Coverage describes how well sequencing reads represent the target region or genome. Good coverage supports stronger data interpretation and more reliable conclusions.
Single-End and Paired-End Sequencing
In single-end sequencing, only one end of the DNA fragment is read. In paired-end sequencing, both ends of the fragment are read. Paired-end sequencing can improve alignment, provide more sequence context, and support more detailed analysis.
Step 4: NGS Data Analysis
The final step is NGS data analysis. Sequencing produces large amounts of raw data, and analysis helps convert that data into useful biological information. NGS data analysis turns sequence reads into meaningful results. The exact analysis workflow depends on the application, such as DNA variant analysis, RNA expression analysis, metagenomics, epigenetics, or targeted sequencing.
Base Calling and FASTQ Files
After sequencing, the instrument identifies each base in each read through base calling. The output is commonly stored in FASTQ files. FASTQ files contain sequence reads and quality scores. These quality scores help show confidence for each base call.
Quality Filtering and Adapter Trimming
Before analysis, reads are processed to prepare clean and organized data for downstream interpretation. Adapter trimming and quality filtering help refine the dataset. This step supports smoother alignment, mapping, and biological analysis.
Demultiplexing
If multiple samples were pooled in one sequencing run, demultiplexing separates reads by their sample-specific indexes or barcodes. This step is essential for multiplexed sequencing because it ensures each sample receives the correct reads.
Alignment and Mapping
In many workflows, filtered reads are aligned or mapped to a reference genome. This helps identify where each read comes from. For applications without a reference genome, de novo assembly may be used to build sequences from overlapping reads.
Variant Calling, Gene Counts, and Interpretation
After mapping, different analysis steps can be performed. In DNA sequencing, variant calling may identify single-nucleotide variants, insertions, deletions, or structural changes. In RNA sequencing, analysis may focus on gene counts, transcript expression, differential expression, or fusion detection.
The final stage is interpretation. This is where researchers connect the data to biological meaning, such as genes, pathways, biomarkers, disease mechanisms, or treatment-related insights.
QC Checkpoints Across the NGS Workflow
Quality control is valuable across the complete NGS sequencing workflow. It helps researchers confirm that each stage is ready for the next one.
Sample QC
Sample QC checks nucleic acid concentration, purity, and integrity before library preparation.
Library QC
Library QC checks library concentration, fragment size distribution, and overall library quality before sequencing.
Sequencing QC
Sequencing QC checks read quality, cluster density, Q-scores, read numbers, and overall sequencing performance.
Data QC
Data QC checks mapping rate, duplication rate, coverage, adapter content, and read quality after processing. These QC checkpoints help researchers maintain confidence from sample preparation to final interpretation.
Choosing the Right NGS Workflow for Your Application
Different research goals require different NGS workflow designs. A workflow for RNA-Seq may not be the same as a workflow for whole genome sequencing or cfDNA analysis.
DNA Sequencing Workflow
DNA sequencing workflows are used for genome sequencing, targeted panels, variant discovery, and mutation analysis. They usually begin with genomic DNA extraction, followed by fragmentation, adapter ligation, sequencing, and variant analysis.
RNA Sequencing Workflow
RNA sequencing workflows begin with RNA extraction. The RNA is usually converted into cDNA before library preparation. RNA-Seq is commonly used for gene expression analysis, transcript discovery, fusion detection, and regulation studies.
cfDNA Workflow
cfDNA workflows work with low-input and naturally fragmented nucleic acids. These workflows benefit from careful extraction, sensitive quantification, and optimized library preparation.
Single-Cell Sequencing Workflow
Single-cell sequencing workflows study genetic or transcriptomic information from individual cells. These workflows require careful sample handling and sensitive preparation methods because the starting material is very limited.
Epigenetics Workflow
Epigenetics sequencing workflows may focus on DNA methylation, chromatin accessibility, or protein-DNA interactions. These workflows may include specialized library preparation and analysis methods.
FireGene supports researchers with NGS Library Prep Kits and related workflow solutions for different sequencing applications, helping laboratories prepare reliable libraries for downstream sequencing.

How to Improve the NGS Workflow
A strong NGS workflow is built with planning, quality control, and application-specific choices. Researchers can improve the workflow by matching the extraction method to the sample type, checking nucleic acid quality, choosing the right library preparation process, and selecting suitable sequencing depth.
Start with the Right Sample
Good sample handling supports good sequencing results. Samples should be collected, stored, and processed in a way that preserves DNA or RNA quality.
Use Suitable Quantification Methods
Accurate quantification helps researchers use the correct input amount for library preparation and sequencing.
Select the Right Library Preparation Strategy
Different applications need different library preparation methods. DNA-Seq, RNA-Seq, cfDNA, single-cell sequencing, and targeted sequencing each have unique workflow needs.
Plan Data Analysis Early
Data analysis should be considered before sequencing begins. Knowing the final research question helps researchers choose the right read length, depth, reference genome, and analysis pipeline.
Applications of NGS Workflow
The next-generation sequencing workflow is used in many research areas. Its flexibility makes it useful for different sample types and scientific goals.
Whole Genome Sequencing
Whole genome sequencing studies the complete genome and is useful for variant discovery, population genetics, and genome-wide analysis.
Targeted Sequencing
Targeted sequencing focuses on selected genes or regions. It is useful when deeper coverage is needed for specific targets.
RNA Sequencing
RNA-Seq helps researchers study gene expression, transcript changes, and regulation patterns.
Single-Cell Sequencing
Single-cell sequencing reveals differences between individual cells, making it valuable for cancer research, immunology, developmental biology, and cell biology.
cfDNA and Liquid Biopsy Research
cfDNA sequencing is used in liquid biopsy research, oncology studies, prenatal research, and other applications where fragmented DNA from biofluids is analyzed.
Metagenomics
Metagenomic sequencing helps study microbial communities from environmental, clinical, or biological samples.

NGS for Beginners: Simple Workflow Summary
For beginners, the NGS workflow can be understood in four simple steps. First, DNA or RNA is extracted from the sample. Second, the extracted nucleic acid is prepared into a sequencing-ready library. Third, the library is sequenced using an NGS platform. Fourth, the generated reads are analyzed with bioinformatics tools to produce meaningful results.
The key to success is the complete workflow. Good extraction, strong library preparation, suitable sequencing design, and careful NGS data analysis all contribute to reliable results.
FAQs
What are the main NGS workflow steps?
The main NGS workflow steps are nucleic acid extraction, library preparation, sequencing, and data analysis. Some workflows may also include target enrichment, library quantification, and multiple QC checkpoints.
What is nucleic acid extraction for NGS?
Nucleic acid extraction for NGS is the process of isolating DNA or RNA from a biological sample. The extracted nucleic acid should have a suitable yield, purity, and quality for library preparation.
What happens during the NGS library preparation process?
During the NGS library preparation process, DNA or cDNA is fragmented, adapters are added, indexes may be included, and the final library is cleaned, quantified, and prepared for sequencing.
What is sequencing by synthesis technology?
Sequencing by synthesis technology reads DNA by detecting nucleotides as they are added to a growing DNA strand. It allows many DNA fragments to be sequenced at the same time.
Why is NGS data analysis important?
NGS data analysis is important because sequencing reads need to be processed, organized, aligned, and interpreted before they can provide useful biological insights.
Is NGS easy for beginners to understand?
Yes. NGS becomes easier to understand when the workflow is divided into clear steps: sample extraction, library preparation, sequencing, and data analysis.
Conclusion
The NGS workflow is a powerful process that transforms biological samples into meaningful genomic information. Each step, from nucleic acid extraction to NGS data analysis, plays an important role in the final result.
For reliable sequencing outcomes, researchers should focus on sample quality, library preparation accuracy, sequencing design, and bioinformatics interpretation. When these steps are planned carefully, next-generation sequencing becomes a practical and highly valuable tool for modern life science research. With the right workflow strategy and reliable reagents, laboratories can move smoothly from sample to sequencing-ready libraries and from raw data to useful biological insights.





